
SUMMARY
OF
SAFETY
AND
EFFECTIVENESS
DATA
(SSED)
I.
GENERAL
INFORMATION
Device
Generic
Name:
Drug-Eluting Coronary
Stent System
(NIQ)
Device
Trade
Name:
IONTM
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent
System
(MonorailM)
IONTM
Paclitaxel-Eluting
Platinum Chromium
Coronary
Stent
System
(Over-The-Wire)
Applicant's
Name
and
Address: Boston
Scientific
Corporation
One
Scimed
Place
Maple Grove,
MN
55311
Date(s)
of Panel Recommendation:
None
Premarket
Approval
Application
(PMA)
Number:
P
100023
Date
of
FDA
Notice
of
Approval:
April 22,
2011
Expedited:
Not
Applicable
II.
INDICATIONS
FOR
USE
The
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary Stent System (Monorail
and
Over-The-Wire
Systems)
is
indicated
for
improving
luminal
diameter
for
the
treatment
of
de
novo
lesions
in
native coronary
arteries
>
2.25
mm to <
4.00
mm
in
diameter
in
lesions
<
34
mm
in
length.
III.
CONTRAINDICATIONS
Use
of
the
ION
Paclitaxel-Eluting Platinum
Chromium
Coronary
Stent
System
is
contraindicated
in
patients
with:
*
Known
hypersensitivity
to
316L
stainless
steel
or
platinum
*
Known
hypersensitivity
to
paclitaxel
or
siructurally
related
compounds
*
Known hypersensitivity
to the
polymer
or
its
individual
components
(see
details
in
Section
4
-
Product
Description
below)
Coronary
Artery
Stenting
is
contraindicated
for
use
in:
*
Patients who
cannot
receive
recommended
antiplatelet
and/or anticoagulant
therapy
*
Patients
judged
to
have
a
lesion
that
prevents complete
inflation
of
an
angioplasty
balloon
or
proper
placement
of
the
stent
or
delivery
device
PMA P100023:
FDA
Summary
of
Safety and
Effectiveness
Data
Page
1
IV.
WARNINGS
AND
PRECAUTIONS
The
warnings
and
precautions
can be
found
in
the
ION
Paclitaxel-Eluting
Platinum
Chromium Coronary
Stent
System
Directions
for
Use
(DFU).
V.
DEVICE
DESCRIPTION
The
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent
System
is
a
device/drug
combination
product comprised
of
two
regulated
components:
*
A
device
(Element
Coronary
Stent System)
*
A
drug
(a
formulation
of
paclitaxel
contained
in
a
polymer
coating).
The characteristics
of
the
ION
stent
system
are
described
in
Table
1
below.
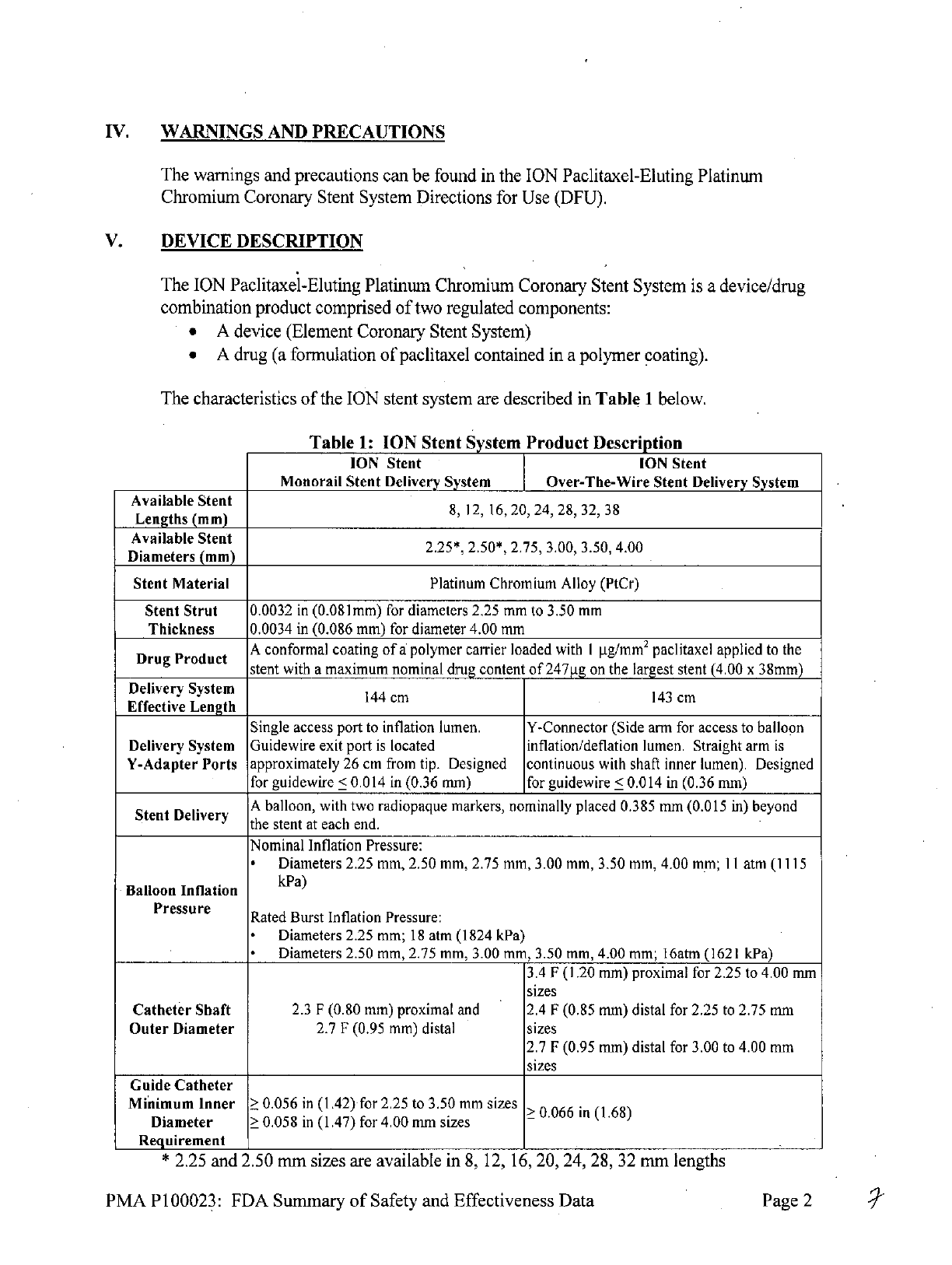
Table
1:
ION
Stent
System
Product
Description
ION
Stent
ION
Stent
Monorail
Stent
Delivery
System
Over-The-Wire
Stent
Delivery
System
Available
Stent
8,
12,
16,
20,
24,
28,
32,
38
Lengths
(mm)
Available
Stent
Dilaete
(m2.25*,
2.50*,
2.75,
3.00,
3.50,
4.00
Diameters
(mm)
Stent
Material
Platinum
Chromium
Alloy
(PtCr)
Stent
Strut
0.0032
in
(0.08
1mm)
for
diameters
2.25 mm
to
3.50
mm
Thickness
0.0034
in
(0.086
mm)
for
diameter
4.00
mm
Drug
Product
A
conformal
coating
of
a
polymer
carrier
loaded
with
I
jig/mm
2
paclitaxel
applied
to
the
stent
with
a
maximum nominal
drug content
of
247pg
on
the largest
stent (4.00
x
38mm)
Delivery
System
143
cm
Effective
Length
Single
access
port
to
inflation
lumen.
Y-Connector
(Side
arm
for
access
to
balloon
Delivery
System Guidewire
exit
port
is
located
inflation/deflation
lumen.
Straight
arm
is
Y-Adapter
Ports
approximately
26
cm
from
tip. Designed continuous
with
shaft
inner
lumen).
Designed
for
guidewire
<
0.014
in
(0.36
mm)
for
guidewire
<
0.014
in
(0.36 mm)
A
balloon,
with
two
radiopaque
markers,
nominally
placed
0.385
mm (0.015
in)
beyond
the
stent
at
each
end.
Nominal Inflation
Pressure:
*
Diameters
2.25 mm,
2.50
mm, 2.75 mm,
3.00 mm, 3.50 mm,
4.00
mm;
11
atm
(1115
Balloon
Inflation
kPa)
Pressure
Rated
Burst
Inflation
Pressure:
*
Diameters
2.25
mm;
18
atm
(1824
kPa)
*
Diameters
2.50 mm,
2.75
mm,
3.00 mm,
3.50
mm,
4.00
mm;
16atm
(1621
kPa)
3.4
F
(1.20
mm)
proximal
for
2.25 to
4.00
mm
sizes
Catheter
Shaft
2.3
F
(0.80 mm)
proximal
and 2.4
F
(0.85
mm)
distal for
2.25 to 2.75
mm
Outer
Diameter
2.7
F
(0.95
mm)
distal
sizes
2.7
F
(0.95
mm)
distal
for 3.00
to
4.00
mm
sizes
Guide
Catheter
Minimum
Inner
0.056
in
(1.42)
for
2.25 to 3.50
mm
sizes
>0.066
in
(1.68)
Diameter
0.058
in
(1.47)
for
4.00
mm
sizes
Requirement
*
2.25
and
2.50
mm
sizes
are
available in
8,
12,
16,
20, 24,
28, 32
mm
lengths
PMA
P100023:
FDA
Summary
of
Safety and
Effectiveness
Data
Page
2

A.
Device
Component
Description
The
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent
System (hereafter
referred
to
as
'ION')
consists
of
a
drug
coated
balloon expandable
stent,
pre-mounted
on
a
high-
pressure
delivery
catheter, similar
to
the
commercially
available
Apex
PTCA
Dilatation
Catheter.
The
stent
is
made
from
a
Platinum
Chromium Alloy
(PtCr).
The
drug
coating
is
composed
of
two
components:
a
polymer
carrier
matrix
material,
poly(styrene-isobutylene-
styrene) referred
to
as
SIBS
(also
known
by
its
commercial name
TransluteTm)
and
an
active
pharmaceutical
ingredient,
paclitaxel.
Each
stent
is
coated
with
1
[ig
paclitaxel per
mm
2
of
stent
surface
area
in
an
8.8%
formulation
(weight
percent
paclitaxel
in
the
polymer
coating). This
is
the
same
dose
density
and
formulation
used
on the
commercially
available
TAXUS®
Express
2
@
Paclitaxel-Eluting
Coronary
Stent System
(P030025) and
TAXUSO
Libert6o
Paclitaxel-
Eluting Coronary
Stent
System (P060008).
PtCr
was
selected
as
the stent
material
as
it
offers
superior strength
and
radiopacity, thus
allowing
the
stent
to
provide
acute
performance
with
thinner
struts
in
a
more
flexible
design.
The
commercial matrix
of
ION
stent
lengths
and
diameters
is
similar
to
the
TAXUS
Express2
and
TAXUS
Libertd
device
matrices. Although
the
matrix
includes
the same
vessel
diameter
range
of
2.25
-
4.00 mm,
ION
has
four
(4)
stent
models
as
compared
to
three
(3)
models
for
TAXUS
Libert6
and
two
(2)
models
for
TAXUS
Express
2.
The
four
(4)
ION
stent
models
are:
*
Small
Vessel
(SV):
2.25
mm
*
Small
Workhorse
(SWH):
2.50
-
2.75
mm
*
Workhorse
(WH):
3.00
-
3.50
mm
*
Large
Vessel
(LV):
4.00mm
The
commercial
matrix
is
shown
in
the
Table
2
below:
Table
2:
ION
Stent
System
Product
Description
A.
D
Stent
Model
Stent
Length
(mm)
u
Tesign
Diameter
(mm)
8
12
16
20
24
28
32
38
C
SV
2.25
x
x
x
x
x
x
x
2.50
x
x
x
x
x
x
x
0
SWH
m
2.75
x
x
x
x
x
x x
-x
P
WH
3.00
x
x
x
x
x
x
x x
o
3.50
x
x
x
x
x
x
x
x
nLV
4.0
x
x
x x
x
x
x
x
e
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
3

B.
Drug
Component
Description
The
drug
component
of
the
ION
Paclitaxel-Eluting Platinum
Chromium
Coronary
Stent
System
consists
of
paclitaxel
(the active
ingredient)
and
TransluteTM
polymer
carrier
(the
inactive
ingredient).
B3.
Paclitaxel
The
active
pharmaceutical
ingredient.
in
the
ION
stent
is
paclitaxel.
It
is
a
white
powder,
isolated
from
a
spectrum
of
Taxus
species
and
hybrids.
The
chemical
name
of
paclitaxel
is:
Benzenepropanoic
acid,
P-(benzoylamino)-a-hydroxy-,6,12bbis(acetyloxy)-12-
(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12bdodecahydro-4,11-dihydroxy-4a,8,13,13
tetramethyl-5-oxo-7,11-methano-1Hcyclodeca[3,4]benz[1,2-
b]oxet-9-yl
ester,[2a
R-
[2aa,4P,4aP,6,9a
(aR*,PS*),
11
a,
1
2a,
12aa,
1
2ba]]-.
Paclitaxel
is
a
diterpenoid
with
a
characteristic
taxane
skeleton
of
20
carbon
atoms,
a
molecular
weight
of
853.91
g/mol
and a
molecular formula
of
C
47
H
51
NO
14
.
It
is
highly
lipophilic,
insoluble
in water,
but
freely
soluble
in
methanol,
ethanol,
chloroform,
ethyl
acetate,
and
dimethyl sulfoxide.
The chemical
structure
of
paclitaxel
is
shown
in
Figure
1.
The
nominal
total
loaded
dose
of
paclitaxel per
nominal
stent
length/diameter
is
shown
in
Table
3
0_
0
-
0
OH
l~J~%..No
....
H17C
4H3
Figure
1:
Chemical
Structure
of
Paclitaxel
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
4

Table
3:
Nominal
Total
Loaded
Dose
of
Paclitaxel
per
Nominal
Stent
Length/Diameter
Nominal
Stent
Length
(mm)
Stent
Model
/
Nominal
S
on
a
e
8
12 16
20 24
28
32
38
Stent
Length
(mm)
SV
39
58
74
94
109
129
148
(2.25
mm)
SWH
Total
loaded
(2.50-2.75
40
62
80
97
115
133
155
181*
dose
mm)
Paclitaxel/Stent
WH
(g)
(3.00-3.50
43
61
86
104
123
141
166
197
mm)
1
1
LV
S(.m
57
82
107
131
156
181
206
247
(4.00
mm)
*
Applies
to
2.75
mm
diameter
only.
B2.
Inactive
Ingredients
The
only inactive
ingredient
in
the
ION
stent
is
SIBS
[poly(styrene-b-isobutylene-b-
styrene)],
a
tri-block
copolymer
(trade
name:
TransluteTM)
that
is
composed
of
styrene
and
isobutylene units
built
on
1,3-di(2-methoxy-2-propyl)-5-tert-butylbenzene.
It
is
a
hydrophobic elastomeric
copolymer
with
a
molecular weight
(Mn-number
average
molecular weight)
of
80,000 to
130,000
g/mol
and
a
polydispersity
index
of
1.0
to 2.0.
The
polymer
is
mixed
with
the
drug paclitaxel
and
then applied
to
the
stents.
There
is
no
primer
or
topcoat
layer.
The
drug/polymer
coating
is
adhered
to the
entire
surface
(i.e.,
luminal
and
abluminal)
of
the
stent.
The
structural
formula
for
the
polymer
is
shown
in
Figure
2
below.
n
ri
rn
=
repeating
units
of
styrene
n =
repeating
units
of
isobulylene
Figure
2:
The
Chemical
Structure
of
Translutem
Polymer
Carrier
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
5

C.
Mechanism
of
Action
The
mechanism
(or
mechanisms)
by
which
an
ION
stent
affects
neointimal
production
as
seen
in
clinical
studies
has
not
been
fully
established.
Paclitaxel
promotes
the
assembly
of
microtubules
from
tubulin
dimers
and
stabilizes
microtubules
by
preventing
depolymerization.
This
stability
results
in
the
inhibition
of
the
normal
dynamic
reorganization
of
the
microtubule
network
that
is
essential
for
vital
interphase
and
mitotic
cellular
functions.
VI.
ALTERNATIVE
PRACTICES
AND
PROCEDURES
There
are
several
other
alternatives
for
the
treatment
of
patients
with coronary
artery
disease,
which
may
include
exercise, diet, smoking
cessation,
drug
therapy, percutaneous
coronary
interventions
(such
as
angioplasty
and
placement
of
bare metal
stents,
coated
stents,
and
other
drug-eluting
stents), and
coronary
artery
bypass
graft surgery
(CABG).
Each
alternative
has
its
own
advantages
and
disadvantages.
A
patient
should
fully
discuss
these
alternatives
with
his/her physician
to
select the
method that
best
meets
expectations
and
lifestyle.
VII.
MARKETING
HISTORY
The
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent System
is
commercially
available
in
the
following countries:
*
Australia
*
Liechtenstein
*
Austria
*
Lithuania
*
Belgium
*
Luxembourg
*
Bulgaria
*
Malaysia
*
Chile
*
Malta
*
Cyprus
0
Netherlands
*
Czech
Republic
*
Norway
*
Denmark
*
Poland
*
Estonia
*
Portugal
*
Finland
*
Romania
*
France
*
Russia
*
Germany
*
Saudi
Arabia
*
Great
Britain
*
Singapore
*
Greece
*
Slovakia
*
Hungary
a
Slovenia
*
Iceland
*
South
Africa
*
India
a
Spain
*
Ireland
*
Sweden
*
Italy
*
Switzerland
*
Latvia
*
Tunisia
*
Lebanon
*
United
Arab
Emirates
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
6

As
of
March
31,
2011,
approximately
29,257
ION
Stents
have
been
distributed
outside
the
U.S.
No
products
have
been
withdrawn
from
the
market
in
any
country
for
any
reason.
VIII.
SUMMARY
OF
PRECLINICAL
STUDIES
A
series
of
non-clinical
laboratory
studies
were
performed
to
evaluate:
*
the
stent
and
the
stent
delivery
system
[i.e.,
the
stent
on
either
the
Monorail
(MR)
or
Over-The-Wire
(OTW) stent
delivery
system
(SDS)]
*
the
polymer
substance
[i.e.,
poly(styrene-isobutylene-styrene)
(SIBS)]
*
the
drug
substance
(i.e.,
paclitaxel)
*
the
finished
combination
product
(i.e.,
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary Stent)
A.
Biocompatibility Studies
A
series
of
Good
Laboratory
Practice
(GLP)
biocompatibility
tests
were
conducted
to
demonstrate
that
the
components
of
the
IONTM
Paclitaxel-Eluting
Platinum Chromium
Coronary
Stent
System (Monorail
and
Over-The-Wire)
are
biocompatible.
Testing
was
conducted
separately
for
the
stent
implant
and
the
stent
delivery
system.
Tests
were
conducted
on
ethylene
oxide-sterilized
bare
metal
(Platinum Chromium
Alloy
(PtCr))
stents,
and
stent delivery systems. These
test
articles
were
processed
in
a
manner
similar
to
the
finished
IONTM
product.
There were
some
minor
manufacturing
differences
which
were
determined not
to
impact
the
biocompatibility
of
the final
device.
Biocompatibility
data
on
the drug
substance, Paclitaxel,
and
SIBs
coating was
incorporated
by
reference
to
P060008
for the
Taxus
Liberte
Paclitaxel-Eluting
Coronary
Stent
System
and
is
applicable
to
the
IONTM
Paclitaxel-Eluting Platinum
Chromium
Coronary
Stent System
because
the
coating process,
amount
of
coating
per
unit
area,
drug
substance
total dose
and
dose
per
unit
area,
and
sterilization
processes
are
equivalent
for the
Liberte
and
ION
stents.
All
biocompatibility testing
was
conducted
in
accordance
with:
*
Guidance
for Industry
and
FDA
Staff:
Non-Clinical
Tests
and
Recommended
Labeling
for
Intravascular
Stents
and
Associated
Delivery Systems,
January
13,
2005
*
Draft Guidance for
Industry:
Coronary Drug-Eluting
Stents
-
Nonclinical
and
Clinical Studies
Companion Document,
March
2008
*
Draft Guidance
for
Industry:
Coronary Drug-Eluting
Stents
-
Nonclinical
and
Clinical Studies,
March
2008
*
Good
Laboratory
Practices
Regulations
(21
CFR
§
58)
*
ISO
10993-1,
Biological
Evaluation
of
Medical Devices:
Evaluation
and
Testing
(2003)
The
biocompatibility
studies
are
summarized
in
Table
4.1
and
4.2.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
7
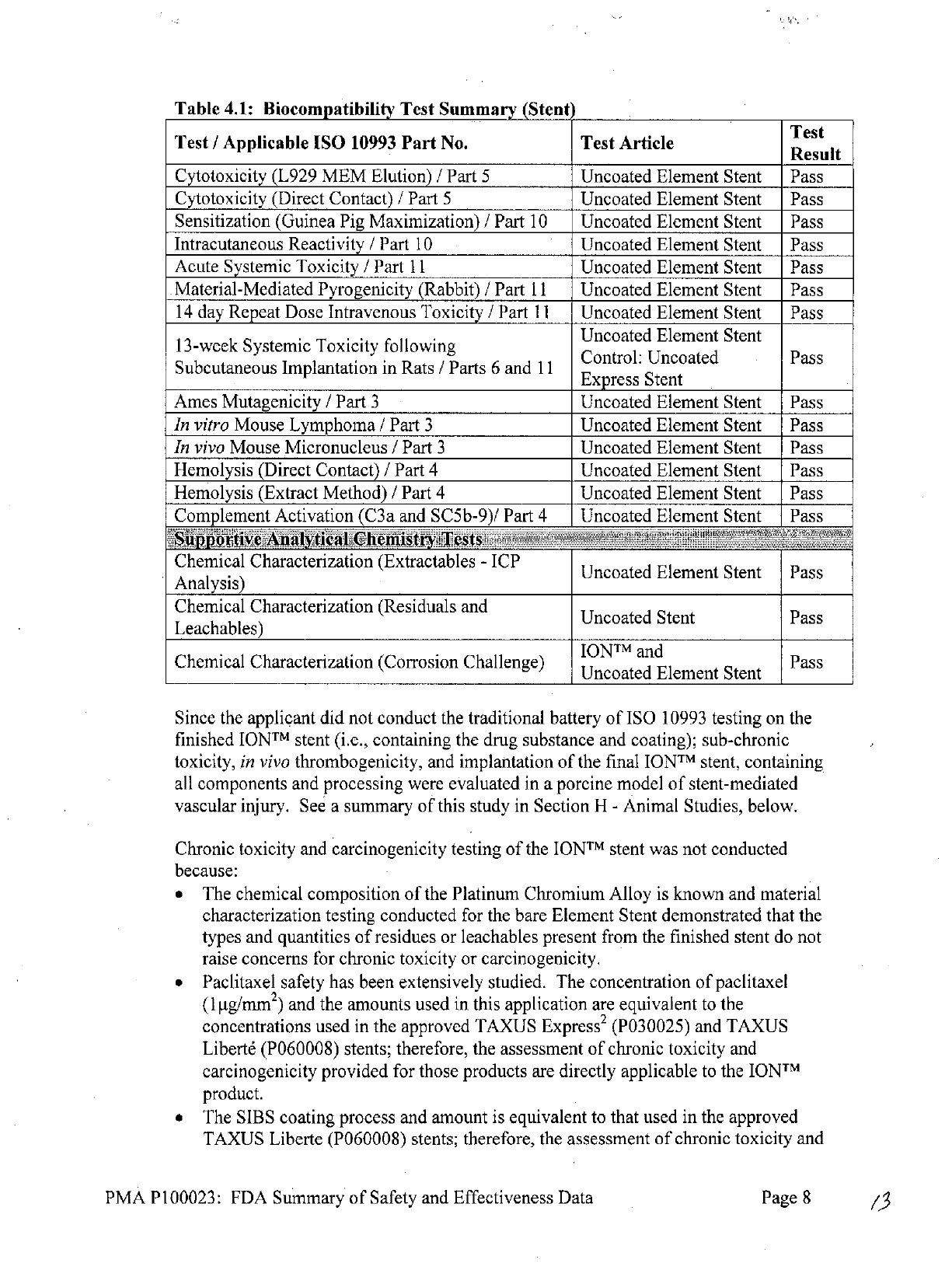
Table
4.1:
Biocompatibility
Test
Summary
(Stent
Test
/
Applicable
ISO
10993
Part
No.
Test Article
Test
Result
Cytotoxicity
(L929
MEM
Elution)
/
Part
5
Uncoated
Element
Stent
Pass
Cytotoxicity
(Direct
Contact)
/
Part
5
Uncoated
Element
Stent
Pass
Sensitization
(Guinea
Pig
Maximization)
/
Part
10
Uncoated
Element
Stent Pass
Intracutaneous
Reactivity
/
Part
10
Uncoated
Element
Stent
Pass
Acute
Systemic
Toxicity
/
Part
11
Uncoated
Element
Stent
Pass
Material-Mediated
Pyrogenicity
(Rabbit)
/
Part
11
Uncoated
Element
Stent
Pass
14
day
Repeat
Dose
Intravenous
Toxicity
/
Part
11
Uncoated
Element
Stent
Pass
13-week
Systemic
Toxicity
following
Uncoated
Element
Stent
13-weekControl:
Uncoated
Pass
Subcutaneous
Implantation
in
Rats
/
Parts
6
and
11
Exprss
Ute
Express
Stent
Ames
Mutagenicity
/
Part
3
Uncoated Element
Stent
Pass
In
vitro
Mouse
Lymphoma
/
Part
3
Uncoated
Element
Stent
Pass
In
vivo
Mouse
Micronucleus
/
Part
3
Uncoated
Element
Stent
Pass
Hemolysis
(Direct
Contact)
/
Part
4
Uncoated
Element
Stent
Pass
Hemolysis
(Extract Method)
/
Part
4
Uncoated Element
Stent
Pass
Complement
Activation
(C3a and
SC5b-9)/
Part
4
Uncoated Element
Stent
Pass
Supportive
Analytical
Chemistry
Tests
Chemical
Characterization
(Extractables
-
ICP
Uncoated
Element
Stent
Pass
Analysis)
Chemical
Characterization
(Residuals
and Uncoated
Stent
Pass
Leachables)
IONTM
and
Chemical
Characterization
(Corrosion
Challenge)
Unoad
Pass
Uncoated
Element
Stent
Since
the
applicant
did
not conduct
the
traditional
battery
of
ISO
10993
testing
on
the
finished
IONTM
stent
(i.e.,
containing
the
drug substance
and
coating);
sub-chronic
toxicity, in
vivo
thrombogenicity,
and
implantation
of
the
final
IONTM
stent,
containing
all
components
and
processing
were
evaluated
in
a
porcine
model
of
stent-mediated
vascular injury.
See
a
summary
of
this
study
in
Section
H
-
Animal Studies,
below.
Chronic
toxicity
and
carcinogenicity testing
of
the
IONTM
stent
was
not
conducted
because:
*
The
chemical
composition
of
the
Platinum Chromium Alloy
is
known
and
material
characterization testing
conducted
for the
bare
Element
Stent
demonstrated
that the
types
and
quantities
of
residues
or
leachables
present
from the
finished stent
do
not
raise
concerns for chronic
toxicity
or
carcinogenicity.
*
Paclitaxel
safety
has
been
extensively
studied.
The
concentration
of
paclitaxel
(1
tg/mm
2
)
and
the
amounts
used
in
this
application
are
equivalent
to
the
concentrations
used
in
the
approved
TAXUS
Express
2
(P030025)
and
TAXUS
Libert6
(P060008) stents;
therefore,
the
assessment
of
chronic
toxicity
and
carcinogenicity
provided
for
those
products
are
directly
applicable
to
the
IONTM
product.
*
The
SIBS
coating
process
and
amount
is
equivalent
to
that
used
in
the
approved
TAXUS
Liberte (P060008)
stents;
therefore,
the
assessment
of
chronic
toxicity
and
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
8

carcinogenicity
provided
for
that
product
is
directly applicable
to
the
IONTM
product.
Table
4.2:
Biocompatibility
Test
Summary
(Deliv
ry
Systems)
Test
Test
/
Applicable
ISO
10993
Part
No.
Test Article
Rest
Result-
.
IONTM
MR
and
OTW
SDS
Cytotoxicity
(L929
MEM
Elution)
/
Part
5
(Cattr
p
nt
Pass
(Catheter
plus
stent)
PROMUS
Element
MR
Pass
Cytotoxicity
(Direct
Contact)
/
Part
5
SDS
(Catheter
only)'
ION
TM
OTW
SDS
Pass
(Catheter
only)
Sensitization
(Guinea
Pig
Maximization)
/
Part
10
ION
TM
MR
and
OTW
SDS
Pass
(Catheter
plus
stent)
Intracutaneous Reactivity
/
Part
10
IONTMMR
and OTW
SDS
Pass
(Catheter plus stent)
Acute Systemic
Injection
/
Part
11
ION
TM
MR and OTW
SDS
Pass
(Catheter plus
stent)
Material-Mediated Pyrogenicity (Rabbit)
/
Part
11
IONTMMR
and
OTW
SDS
Pass
(Catheter plus
stent)
Hemolysis (Direct Contact
/
Part
4
ION
TM
MR
and
OTW
SDS
Pass
(Catheter plus
stent)
PROMUS
Element
MR
Pass
.
SDS
(Catheter
only)'
Hemolysis
(Extract)
/
Part
4 SDS
IONTM
OTW
SDS
Ps
(Catheter
only)
Complement
Activation
(C3a and
SC5b-9)
/
Part
4
ION
TM
MR
and OTW
SDS
Pass
(Catheter
plus stent)
Supportive
Analytical
Chemistry
Tests
.
.110T
MR
and OTW
SDS
USP
Physicochemical
Test
for
Plastics
/
Part
18
j
MR
p
nt ]
Pass
(Catheter plus
stent)
'The
PROMUS
Element
and
IONTM
Monorail
delivery
catheters
consist
of
the
identical
materials
and
similar
processing,
and
data
provided
support
that
any
differences
will not
affect
the
biocompatibility
of
the
final
product.
Therefore
the
data
from
the
PROMUS
Element
Monorail
testing
are
applicable
to
the
IONTM
Monorail
device.
The
applicant
did not
conduct traditional in
vivo
thrombogenicity
on the
IONTM
MR
and
OTW
delivery
systems.
The
potential
for
thrombogenicity
was
evaluated
in
a
porcine model
of
stent
mediated
vascular
injury.
See
a
summary
of
this
study
in
Section
H
-
Animal studies, below.
Use
of
the
vascular implant
study
in
the
porcine
model
was
deemed acceptable because
the
materials
of
manufacture,
design,
and
processing
methods
for
the delivery
system
are
equivalent
to
the
approved
Apex
balloon
catheter
(P860019/SO28).
The
handle
of
this
delivery
system
incorporates
nanotechnology.
Detailed
manufacturing
information
and
material
characterization
and
biocompatibility testing
were
provided
to
confirm
that
there
is
no
release
of
nanoparticles
from
the
handle
of
this
device.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
9

Based
on the
testing
performed
for the
Element
stent
and
delivery
systems,
as
well
as
the
established
biocompatibility
and
safety data
on
paclitaxel
and
SIBS, it
can
be
concluded
that
IONTM
Coronary
Stent
System
is
biocompatible
for
its
intended
use.
B.
In
Vivo
Pharmakokinetics
B1.
ION
Paclitaxel-Eluting
Platinum Chromium
Coronary
Stent
Boston
Scientific has
provided
a
letter
from
the
drug
substance
manufacturer
authorizing
access
to
a
Drug Master
File
(DMF)
in
support
of
this
application.
The
drug
substance
manufacturer
produces
a
generic
form
of
the
drug
Taxol
,
a
Bristol
Myers
Squibb
drug
product that
is
approved
for
injection
of
multiple oncologic
indications.
In
vivo
animal
and
in
vitro
pharmacology
and
toxicology
studies,
as
well
as
in
vivo
animal
and
human
pharmacokinetic
studies,
were
conducted
on Taxol
to
provide
information
about
systemic, regional
and
local
toxicity, dose-related toxicity,
distribution
profiles,
end-
organ
disposition,
drug
metabolism,
and
potential drug-drug interactions.
Given
that
the
polymer
coating
and drug
component
of
the
ION
is
identical
to
that
of
the
TAXUS
Express
2
(P030025)
and
TAXUS
Libert6
(P060008), the
evaluation
of
TAXUS
Express
2
and
TAXUS
Libert6
is
applicable.
In
the
clinical
studies
TAXUS
1,
11,
and
III
(which evaluated
TAXUS8 Express
2
@)
and
ATLAS
(which
evaluated
TAXUS
Libert6),
no
paclitaxel
levels
were
detected
after
stent
implantation
using
an
analytical
method
with
a
lower limit
of
quantification
(LLOQ)
of
10
ng/ml.
These
findings
were
confirmed
in
preclinical studies using
multiple
stents
with
total
loaded doses above
the
clinically available
stent
system
and
an
assay
with
an
LLOQ
of
0.03
ng/ml.
Hence,
in
the
absence
of
systemically
detectable
levels,
standard
pharmacokinetic
parameters
were
not established.
B2.
Drug
Interactions
Paclitaxel
is
metabolized
in
the
liver via
CYP2C8
to
6-alpha-hydroxypaclitaxel
and
via
CYP3A4
to
3'-p-hydroxypaclitaxel
and
6-alpha,
3'-p-dihydroxypaclitaxel.
Paclitaxel
is
a
substrate
of
P-glycoprotein.
Because
metabolism
appears
to
play
an
important
role
in
the
elimination
of
paclitaxel,
agents
that
could compete
with
or
inhibit
the
CYP2C8 and
CYP3A4 isoenzymes
may
increase
paclitaxel
plasma
levels.
Potential
drug
interactions
may occur
with
any
drug
that
affects
these
isoenzymes.
Formal
drug
interaction
studies have not been
conducted with
the
ION
stent.
Consideration should
be
given
to
the
potential
for
both
systemic and
local
drug
interactions
in
the
vessel
wall
when deciding
to
place
an ION
stent
in
a
patient
who
is
taking
a
drug
with known
interactions
to
paclitaxel
or
when
deciding
to
initiate
therapy
with
such
a
drug in
a
patient
that
has
recently received
an
ION
stent.
C.
In
Vitro
Engineering
Testin2
In
vitro
engineering
testing
on
the
ION
Stent
System
was
conducted,
as
applicable,
in
accordance
with:
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
10

*
FDA
Guidance
for
Industry
and
Staff:
Non-Clinical
Tests
and
Recommended
Labeling
for
Intravascular
Stents
and
Associated
Delivery
Systems,
January
13,
2005,
*
FDA
Draft
Guidance
for
Industry and
Staff:
Coronary
Drug-Eluting
Stents
-
Nonclinical
and
Clinical
Studies,
March
2008,
and
*
FDA
Guidance
for
Industry
and
Staff:
Establishing
Safety
and
Compatibility
of
Passive
Implants
in
the
Magnetic
Resonance
(MR)
Environment,
August
2008.
In
vitro
engineering
testing
was
performed
on
the
ION
stent
mounted
on
either
the
MR
or
OTW
delivery catheters.
The
in
vitro
engineering
studies
conducted
are
summarized
in
Table
5
"Pass" denotes
that
the
test
results
met
product
specifications
and/or
the
recommendation
in
the above-
referenced
guidance
documents.
Additional
testing was
conducted
to
support
the
integrity
of
the
coating
on
the
IONTM
stent
as
shown
in
Section
IX
D
-
Drug
Coating
Characterization
Testing.
Table
5:
Stent
and
Delivery
Catheter
Engineering
Testing
1
Test
Test Description of Test
Rest
Results
Stent
Dimensional
and
Functional
Attributes
Material
Chemical
analysis
was
conducted
on
the
Platinum
Chromium
Pass
Composition
(PtCr)
ingot
provided
by
the
material
supplier
to
confirm
both
chemical
analysis
and
inclusion/impurity
content
as
provided
by
ASTM
F138-00
"Standard Specification
for
Wrought
18
Chromium-14
Nickel-2.5 Molybdenum Stainless
Steel
Bar and
Wire
for
Surgical
Implants
(UNS
S31673)."
The
analysis
confirmed that
the
chemical
composition
matched
the
ASTM
standard.
Stent
Corrosion
ION
stents were
tested
according
to
ASTM
F2129-01
"Standard
Pass
Resistance
Test Method
for
Conducting
Cyclic
Potentiodynamic
Measurements,"
ASTM
F756,
ASTM
G-71
to
Determine
the
Corrosion
Susceptibility
of
Small
Implant
Devices"
to
demonstrate
that
the
finished
stents
exhibit
acceptable
breakdown,
repassivation,
and
galvanic
coupling corrosion characteristics.
The
results
indicated
that
the
corrosion
resistance met
product specification.
Testing
also
included
an
assessment
of
Fretting
Corrosion
after
pulsatile
fatigue
cycling.
The
results
demonstrated that
there
was
no
evidence
of
fretting
corrosion
with
ION
stents
under
overlapping,
bent,
pulsatile
fatigue
conditions
up
to
400
million
cycles.
Dimensional
To
measure
and
inspect
the
substrate
(uncoated)
Element
stent
to
Pass
Verification document
that
the
un-expanded
stent
dimensional
specifications
meet
the
product
design
requirements.
All
products
met
specifications.
Percent
Surface
Stent
surface
coverage
as
a
function
of
stent
diameter
was
measured
Pass
Area
for the
IONTM
stent.
The
percent
surface
area
is
determined
by
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
11
dividing
the
measured
total
contact
surface
area
of
the
coated
stent
by
the
surface
area
of
the artery
based
on
deployed
stent
measurements
at
the
nominal
stent
diameter.
The
calculated
percent
surface
area
of
the
stent
should
be
<23.9%
when expanded
to
the
lowest
labeled
diameter.
Values
were
calculated
for
2.00
to
4.00
mni stent
diameters.
Foreshortening
The
lengths
of
the
stents
were
measured
prior
to
and
after
Pass
expansion
to
nominal diameter.
All
stents
met
product
specifications.
Recoil
for
Testing
was
conducted
to
quantify
the
amount
of
elastic
recoil
for
Pass
Balloon
the
stent
and
correlate
this
parameter
to
the
recommended
sizing
Expandable
procedures.
Results
indicated
that
product
specifications
were
met.
Stents
Stent
Testing
was
conducted
to
determine
whether
the
deformation
Pass
Overexpansion
experienced
by
the
stent
undergoing expansion
above
the
maximum
rated
diameter
gives rise
to
stent
or
coating
fractures.
No
stent
exhibited
any strut fracture
or
indications
of
coating integrity
issues
when
visually
examined
at
32X
following
over-expansion..
Radial
Stiffness Testing was
conducted
to
determine
the
ability
of
the
IONTM
stent
Pass-
and
Radial
to
resist deformation
under
radial
loads.
Strength
Compression
Testing
was
conducted
to
determine
the
radial
resistance
of
the
Pass
Resistance
IONTM
stent
to
external
compression.
Mechanical
Ultimate tensile
strength, yield strength
and
elongation
testing
was
Pass
Properties performed
on
tubing (pre-processing)
used
to
fabricate
the
stents.
Ultimate tensile
strength,
yield
strength
and
elongation
on
pre-
processed tubing
met
product specification. Analysis
of
SEM
images
on
stent
components
at
various
process
stages
determined
that
mechanical properties
were
not altered
by
processing.
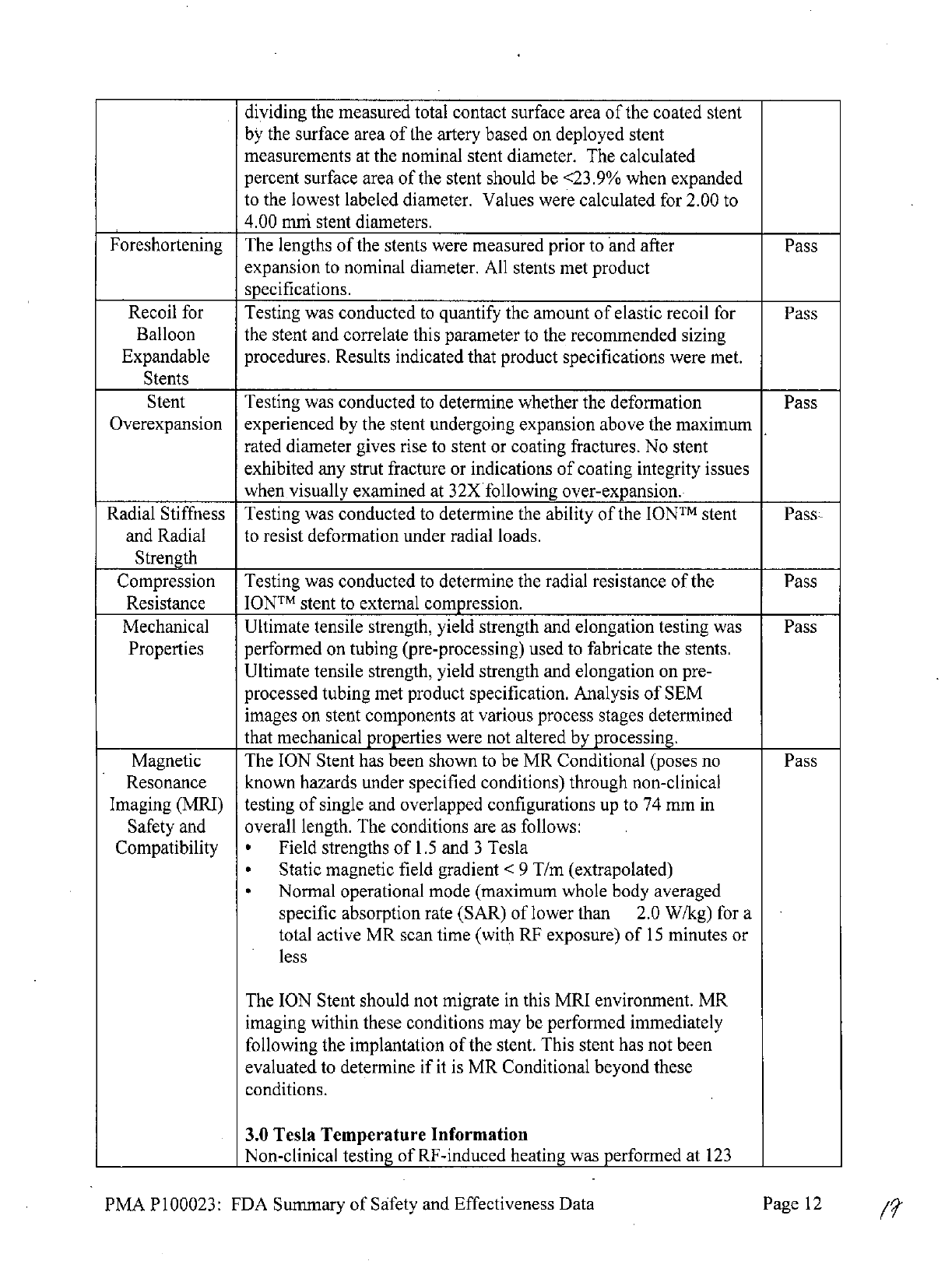
Magnetic
The
ION
Stent
has
been shown
to
be
MR
Conditional
(poses
no
Pass
Resonance
known
hazards under
specified
conditions) through non-clinical
Imaging
(MRI)
testing
of
single
and
overlapped configurations
up
to
74
mm
in
Safety
and
overall
length.
The
conditions
are
as
follows:
Compatibility
*
Field
strengths
of
1.5
and
3
Tesla
*
Static
magnetic
field
gradient
<
9
T/m
(extrapolated)
*
Normal
operational
mode
(maximum
whole
body
averaged
specific
absorption
rate
(SAR)
of
lower
than
2.0
W/kg)
for
a
total
active
MR
scan
time
(with
RF
exposure)
of
15
minutes
or
less
The
ION
Stent should
not
migrate
in
this
MRI
environment.
MR
imaging
within
these
conditions
may
be
performed
immediately
following
the
implantation
of
the
stent.
This
stent
has
not
been
evaluated
to
determine
if
it is
MR
Conditional
beyond
these
conditions.
3.0
Tesla
Temperature
Information
Non-clinical
testing
of
RF-induced
heating
was
performed
at
123
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
12

MHz
in
a
3.0
Tesla
Magnetom
Trio®,
Siemens
Medical
Solutions
MR
system, software
version Numaris/4,
Syngo®
MR
A30.
RF
power
was
applied
for
15
minutes
and
the
measured conductivity
of
the
phantom
material
was about
0.3
S/m.
The
phantom
average
SAR was
calculated
using
calorimetry
to be 2.2
W/kg.
The
maximal
in-vitro temperature
rise
was
calculated
as
2.6
0
C
for
a
measured
stent
length
of
74
mm
with
the
whole-body
SAR
scaled
to
2.0
W/kg.
The
calculations
did
not
include
the
cooling
effects
due to
blood
flow.
1.5
Tesla
Temperature
Information
Non-clinical
testing
of
RF-induced heating
was
performed
at
64
MHz
in
a
1.5
Tesla
Intera®
Philips
Medical
Systems,
software
version
Release
10.6.2.0,
2006-03-10
whole
body
coil
MR scanner.
RF
power
was applied
for
15
minutes
and
the
measured
conductivity
of
the
phantom
material
was
about
0.3
S/m.
The
phantom
average
SAR
was
calculated
using
calorimetry
to
be
2.1
W/kg.
The
maximal
in-vitro
temperature
rise
was calculated
as
2.6
0
C
for
a
measured
stent length
of
74
mm
with
the
whole-body
SAR scaled
to
2.0
W/kg.
The
calculations
did
not
include
the
cooling effects
due to
blood
flow.
In
vivo,
local
SAR depends on MR Field
strength
and
may
be
different
than
the
estimated
whole body averaged
SAR,
due
to
body
composition,
stent
position
within
the
imaging
field,
and
scanner
used,
thereby
affecting
the
actual
temperature
rise.
Image
Artifact
Information
The
calculated
image
artifact extends
approximately
7
mm
from
the
perimeter
of
the
device
diameter
and
5
mm beyond
each
end
of
the
length
of
the
stent when
scanned
in
non-clinical
testing
using
a Spin
Echo
sequence.
With
a
Gradient
Echo
sequence
the
calculated
image
artifact
extends
5
mm
beyond
the
perimeter
of
the
diameter
and
6
mm
beyond
each
end
of
the
length with
both sequences
partially
shielding
the
lumen
in
a
3.0
Tesla Intera (Achieva
Upgrade),
Philips
Medical
Solutions,
software
version
Release
2.5.3.0
2007-09-28
MR system with
a
transmit/receive
head
coil.
Medical
Registration
It
is
recommended
that
patients
register
the
conditions
under
which
the
implant
can
be
scanned
safety
with
the
MedicAlert Foundation
(www.medicalert.org)
or
equivalent
organization.
Radiopacity
Testing
was
conducted
on
the
bare
metal
stent
as
the
addition
of
the
Pass
coating
did not
add
or
detract
from
the
radiopacity
of
the
stent
in
clinical
use.
Stent
Delivery
System
Dimensional
and
Functional
Attributes
Delivery,
The
delivery,
deployment
and
retraction
of
the
IONTM
Stent System
Pass
Deployment
was
assessed
by
testing
system
track, crossing
profile,
stent
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
13
and
Retraction
deployment,
guidewire
movement
and
balloon
withdrawal
from
a
stent
and into
the guide
catheter. Testing
demonstrated that
the
IONTM
stent
system
could
be
delivered
to
the
target
location,
deployed,
and
retracted,
thus
meeting required
product
specifications.
Balloon
Rated
IONTM
Stent Systems
were
tested
to
failure
to
demonstrate that
the
Pass
Burst Pressure
stent system met rated
burst
pressure.
All
stent
systems
met
(RBP)
specification
and
demonstrated
with
95%
confidence
that
at
least
99.9%
of
balloons
will
not experience loss
of
integrity
at
or below
the
rated
burst pressure.
Balloon
Fatigue
IONTM
Stent Systems across
the range
of
stent/balloon
lengths
and
Pass
diameters
were
required
to
complete
10
pressurization
cycles
to
Rated
Burst
Pressure
(RBP).
The
results
show
statistically
that,
with
95%
confidence,
90%
of
the
catheters
will
not
experience
balloon,
shaft,
or
proximal/distal
seal
loss
of
integrity
at
or
below
the
maximum
recommended
rated
balloon burst pressure.
Stent
Diameter
Testing
was
performed
to
determine how
the
diameter
of
a
deployed
Pass
vs.
Balloon stent
varies
with applied
balloon
pressures.
The
stent
sizing
results
Pressure
verify
that
the
stent
systems
meet
the
labeled
compliance values.
Catheter
Bond
Representative
sizes
of
the
IONTM
stent
delivery system
were
tested
Pass
Strength
to
determine
the
balloon
bond
and
full
unit tensile strength
of
the
delivery
system.
All
stent systems exceeded
the
minimum
specifications
for
full
unit tensile
strength
and
balloon
bond.
Balloon
IONTM
delivery systems
across
the
range
of
balloon
lengths
and Pass
Deflation
Times
diameters
were
tested
for
deflation
times,
and
all
stent
systems
met,
specifications.
Stent
Testing was
conducted
to
assess
the forces
required
to
displace
a
Pass
Securement
for
crimped
IONTM
stent
from
the
delivery
systems
(1)
directly
from
the
Unsheathed
balloon
catheters,
(2)
after tracking
through
a
simulated
tortuous
Stents
artery
model
and
(3)
after
tracking
through
a
simulated tortuous
artery
model
and
then
through
a
simulated lesion.
All
stent systems
met
the
stent
securement specification.
Non-Coaxial Testing consists
of
withdrawing
a
catheter with
a
mounted
stent non-
Pass
Withdrawal
into
coaxially
into
a
simulated
guide
catheter
tip,
at a
45oC
angle,
a
Simulated
following
a
track
conditioning
step,
in
which
the
IONTM
Stent
Guiding
Delivery Systems
were
challenged
by
repeatedly
tracking
the
Catheter
crimped stent system
three
(3)
times
through
a
tortuous
artery
model.
The
unit
was
then
assessed
for
stent
movement.
All
samples met the
product
specification.
Balloon
Testing
was
conducted
to
verify
that
the
IONTM
stent
system
can
be
Pass
Catheter
safely
withdrawn
back into
the
recommended
guide
catheter
sizes
Withdrawal
both
before and
after
stent
deployment.
All
samples met
the
product
Resistance
specification.
Coating
The
coating
durability
of
the
IONTM
stent
coating
was
assessed
via
a
Pass
Durability
series
of
acute
in
vitro
tests
performed
on
the
coated stent
and
the
SIBS
polymer.
The
test
results
demonstrate
that
the
paclitaxel/SIBS
coating displays
good
durability
and
coating
integrity.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
14
Coating
Coating
Adhesion
and
Cohesion
testing
has
been
performed
to
Pass
Adhesion
and
.
assess
the
adhesive
and
cohesive
properties
of
the
IONTM
stent
Cohesion
coating.
The
IONTM
coating demonstrates
adequate
adhesion
and
cohesion properties.
The
coating
has
a
high
resistance
to
detachment
from
the
stent
and
is
therefore
considered
acceptable
for
intended
use.
Stress
and
Using Finite
Element
Analysis
(FEA),
stress
and
strain
analysis
was
Pass
Strain
Analysis
performed
on
the
stent
and
the
stent coating
to
demonstrate.
that
they
(Finite
Element
maintain
acceptable
safety
in
stress
loading
environments,
Analysis
simulating
nominal
and
overexpansion,
and
bending
and
radial
(FEA))
conditions.
The
FEA
evaluated
the
structural
integrity
of
the
stent
and
coating
when subjected
to
the
expected
loading
conditions
generated
in
coronary
arteries.
The
analysis
took
into
account
manufacturing,
delivery,
implantation
and
clinical
loading over
the
implant
life,
and
predicted
that
fatigue
failures
will
not
occur
over
400
million
cycles
(10-years)
of
loading.
Accelerated
Accelerated
durability
testing
was
performed
on the
IONTM
stent
and Pass
Durability
the
stent coating
to
demonstrate
that
the
structural integrity
and/or
Testing coating
integrity
is
maintained following
exposure
to
the
pulsatile
stresses
and
strains
exceeding those
typically experienced
by
a
human
coronary
artery
for
10
years (400
million
cycles).
Testing
included
assessment
of
Post-Elution
Fatigue
and
Overlapping
Pulsatile
Fatigue
on
a
Curve
(PFC).
All
tested
stents
were
free from
fatigue
induced
surface defects,
and
there
was
no
evidence
of
coating
fatigue
or
corrosion.
The
stent met the
10
year accelerated
fatigue
resistance
requirement
of
the
product specification.
The
overlapping
stents
were
also
evaluated
in
an
accelerated
in
vitro
test
of
approximately
10
year
equivalent
real
time
and
met
visual
requirements
for coating integrity
and
strut
damage.
Particulate Particulate
testing
has been
performed
on the
IONTM
stent
system
in Pass
Testing accordance
with
the
recommendations
in
the
draft
FDA
Guidance
for
Industry:
Coronary Drug-Eluting
Stents
-
Nonclinical
and
Clinical Studies,
March
2008,
including
revisions presented during
the
2008
Transcatheter Cardiovascular Therapeutics (TCT)
Conference,
Washington
DC,
October
15,
2008.
Testing
included
assessment
of
Baseline Particulate
(Overexpansion), Simulated
Use
Particulate,
and
Chronic Particulate
(Pulsatile
Particulates
in
a
Curve
(PPC)).
The
results demonstrate that
the
particulate
counts
are
similar
or
lower
for
IONTM
coated
stents
compared
to
bare
Element
stents.
Given
comparable particulate
counts, and
that
there
is
no
evident
increase
in
counts
on
IONTM
coated stent
product,
no
chemical
identification
of
particulates
was
warranted.
D.
Drug
Coating
Characterization
Testing
The
coating
characterization
testing
conducted on
the
ION
stent
coating
is
summarized
in
Table
6.
PMA
P100023:
FDA
Summary
of
Safety and
Effectiveness
Data
Page
15

Table
6:
Coating
Characterization
Testing
Test
Description
of
Test
Polymer
components
were
tested
to
ensure
conformity
to
raw
material
Polymer
specifications
and
incoming
inspection
procedures.
The
analysis
confirmed
the
material
met specifications.
Assays
were
conducted
to
determine
Mw,
Mn,
polydispersity,
Chemical
Analysis-
monomer
content,
presence/formation
of
oligomers,
and
free
Polymer
monomers.
The
results
of
each
assay
met
specifications
established
by
the
applicant.
Chemical
Analysis
-
Drug
substance
was
tested
to
ensure
conformity
to
incoming
Certificate
Drug
of
Analysis
(COA);
the
testing
confirmed
conformity
to
the
COA.
Assay
was
conducted
to
quantitatively
determine
the
total
amount
of
Drug
Content
the
drug
substance,
paclitaxel,
on
the
ION
stent.
The
results
verified
that samples
met
the
targeted
drug
content specifications.
Dose
Density
Dose
per
unit
area
was
calculated.
C
n
UTesting
was
conducted
to
verify
the
reproducibility
of
coating
t
uniformity
from
stent
to
stent
and
batch
to
batch.
Testing confirmed
Reproducibility.
.
adequate
coating
uniformity/reproducibility
as
outlined
in USP
<905>.
Assays
were
conducted
to
quantitatively determine
the
type
and
Impurities/Degradation
amount
of
impurities
and
degradation products
on the
ION
stent.
Products
Testing confirmed
acceptable
levels
of
impurities
and
degradation
products.
Assay
was
developed
to
measure
the
in
vitro
release
kinetics
of
In
vitro
Elution
paclitaxel
off
the
ION
stent.
An appropriate
method
and
specifications
were
developed
for
this parameter.
Particulate
levels
were
evaluated
for the
ION
stent system
under
Particulates
simulated
use
conditions,
including
tracking
and
deployment
(see
Table
1
5
above).
E.
Chemistry,
Manufacturing,
and Controls (Quality)
Testing
Samples
from
each
batch
of
finished
stents
are
subjected
to
certain
tests
prior
to
release.
This
testing
is
summarized
in
Table
7.
Where
applicable,
the
test
methods
follow
International Conference
on
Harmonization
(ICH)
Guidelines. Information
to
support
the
stability
of
ION
is
summarized
separately
in
Section
IX
F
-
Stability
below.
Table
7:
ION
Stent
Release
Testing
Test
Description
of
Test
Material
Analysis
-
The
polymer
was
tested
to
ensure
conformity
to
specifications.
The
Polymer
polymer
met
specifications
prior
to
utilization
in
finished
goods.
Assay
is
conducted
to
verify
the
identity
of
the
drug
substance,
paclitaxel,
in
the
ION
stent.
Drug
Assays
are
conducted
to
quantitatively
verify
the
amount
of
drug
and
the
Content/Impurities
type
and
amount
of
impurities
on
the
ION
stent.
Drug
Content
Multiple
stents
are
assayed
to
verify
the
uniformity
of
the
drug
content
Uniformity
between
individual
stents
is
within
specifications
established
for
the
ION
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
16

Table
7:
ION
Stent
Release
Testing
Test
Description
of
Test
stent.
In
vitro
Drug
Elution
The
in
vitro
release
profile
of
paclitaxel
is
measured
to
verify
that
the
drug
release
is
within
the
specifications
established
for
the
ION
stent.
Particulates
Particulate
counts
are
measured
to
verify
that
they
remain
below
acceptable
levels
established
for
the
ION
stent.
F.
Stability
Stability
studies
specific
to
the
polymer/drug
coating
component
were
conducted
in
accordance
with
ICH
guidelines
to
establish
a
shelf
life/expiration
date
for
the
ION
stent
system.
In
addition,
testing
to
establish
package integrity
and
functional
testing
of
the
stent
and
delivery
system
were
conducted
on aged
product
to
ensure that
ION
continues
to
meet
specifications
throughout
its
shelf
life.
The
data
generated
support
a
shelf
life
of
18
months.
Stability studies
included
evaluation
of
drug
identity,
assay,
degradants,
in
vitro
elution, particulates,
sterility,
drug
content uniformity,
residual solvents,
and
endotoxin.
In
addition,
the stability
of
the
drug
substance
and
inactive
polymer
has
been
independently
verified.
G.
Sterilization
The
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent System
(Monorail
and
Over-The-Wire)
is
sterilized
using
ethylene
oxide sterilization and
has
been validated
per
AAMI/ISO
11135:1994 "Medical
Devices
-
Validation
and
Routine
Control
of
Ethylene
Oxide
Sterilization."
Results
obtained
from the
sterilization
studies
show
that
the
product
satisfies
a
minimum
Sterility
Assurance
Level
(SAL)
of
10-6.
The
amount
of
bacterial
endotoxin
was
verified
to
be
within
the
ANSI/AAMI
ST72
specification limit
or
ION
stent
delivery
systems.
H.
Animal Studies
Because
detailed
arterial
histopathology
and
histomorphometry
and
pharmacokinetic
data
cannot
be
obtained
through
human clinical
trials,
a
series
of
animal
studies
were
conducted
to
evaluate safety,
vascular compatibility,
in
vivo drug release, and acute
product
performance.
The
safety,
vascular
compatibility,
and
acute
performance
of
ION
paclitaxel-eluting
stents
were
evaluated
in
the
non-injured
porcine
coronary
artery
model. Studies
were
conducted
in
accordance
with
§21
CFR
58
(Good
Laboratory Practices).
The
consistency
of
the
vascular
response
to
the
Element
platform
with
the
approved
Libert6
platform
was
evaluated
and
compared
in
the
Overlap
ION
Study
1133-024.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
17
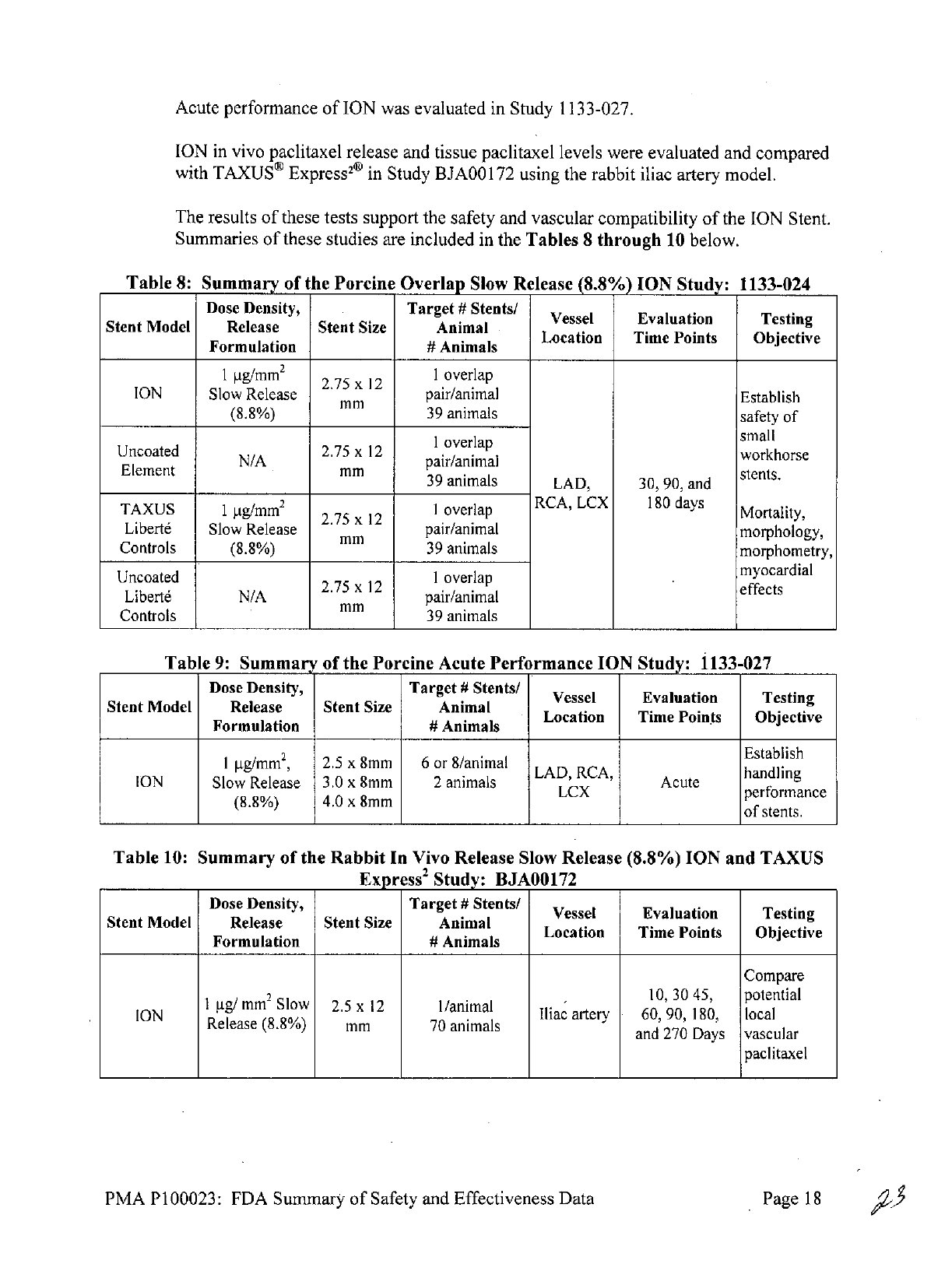
Acute
performance
of
ION
was
evaluated
in
Study
1133-027.
ION
in
vivo
paclitaxel
release
and
tissue
paclitaxel
levels
were
evaluated
and compared
with
TAXUS8
Express20
in
Study
BJAOO
172
using
the
rabbit
iliac
artery
model.
The
results
of
these
tests
support
the
safety
and
vascular
compatibility
of
the
ION
Stent.
Summaries
of
these
studies
are
included
in
the
Tables
8
through
10
below.
Table
8:
Summary
of
the
Porcine
Overlap
Slow
Release
(8.8%)
ION
Study:
1133-024
Dose
Density,
Target
#
Stents/
Vessel
Evaluation
Testing
Stent
Model
Release
Stent
Size
Animal
Formlaton
#Anials
Location
Time
Points
Objective
Formulation
#
Animals
I
ig/mm
2
2.75
x
12
1
overlap
ION
Slow
Release
pair/animal
Establish
(8.8%)
39
animals
safety
of
I
overlap
small
Uncoated
N/A
2.75
x
12
pair/animal
workhorse
Element
NAmm
pair/nima
39
animals
LAD,
30,
90,
and
stents.
TAXUS
1
Ig/mm
2
2.75
x
12
1
overlap
RCA,
LCX
180
days
Mortality,
Libert6
Slow
Release
mm
pair/animal
morphology,
Controls
(8.8%)
39
animals
morphometry,
Uncoated
I
overlap
myocardial
Libert6
N/A
2.75
x
12
pair/animal
effects
Controls
mm
39
animals
Table
9:
Summary
of
the
Porcine
Acute
Performance
ION
Study:
1133-027
Dose
Density,
Target
#
Stents/
Vessel
Evaluation
Testing
Stent
Model
Release
Stent
Size
Animal
Formlaton
UAnials
Location
Time
Points
Objective
Formulation
#
Animals
2
Establish
ION
Slow
Release
3.0
x
8mm
2
animals
h
g
Acute
18
42.5
x
8mm
6or
l
LCX
performance
(8.8%)
4.0
x
8mm
of
stents.
Table
10:
Summary
of
the
Rabbit
In
Vivo
Release
Slow
Release
(8.8%)
ION
and
TAXUS
Express
2
Study:
BJA00172
Dose
Density,
Target
#
Stents/
Vessel
Evaluation Testing
Stent
Model
Release
Stent
Size
Animal
Formlaton
UAnials
Location
Time
Points
Objective
Formulation
#
Animals
Compare
1
ig/
mm
2
Slow
2.5
x
12
1/animal
10,3045,
potential
ION
RIliac
artery
60, 90,
180,
local
Release
(8.8%)
mm
70
animalsan27Dysvcur
and 270
Days
vascular
paclitaxel
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
18

Dose
Density,
Target
#
Stents/
Vessel
Evaluation
Testing
Stent
Model
Release
Stent
Size
Animal
Formlaton
#Anials
Location
Time Points
Objective
Formulation
#
Animals
exposure
for
SR
Element
versus
SR
TAXUS
2.5
x
12
1/animal
Express
2
Express
2
mm
70
animals
using
stent
content
and
tissue
paclitaxel
levels.
IX.
SUMMARY
OF
PRIMARY
CLINICAL
STUDIES
The
applicant
collected
clinical data
through
the
PERSEUS
Clinical
Trial
Program,
to
establish
a
reasonable
assurance
of
safety
and
effectiveness
of
coronary
artery
stenting
with
the
ION
stent
for
improving
luminal diameter
for the
treatment
of
de
novo lesions.
The
PERSEUS
program
consisted
of
two
(2)
parallel studies,
PERSEUS
Workhorse
(WH) and
PERSEUS
Small
Vessel
(SV).
The
PERSEUS
WH
included
sites
in
the
United
States,
New Zealand,
Australia,
and
Singapore.
The
PERSEUS
SV
was
a
US-
only study. Both
trials
were
evaluated
under
IDE
G060237.
Data
from
this clinical
study
were
the
basis
for
the
PMA
approval
decision.
A
summary
of
the
clinical
study
is
presented
in
Table
11
below.
PERSEUS WH
The
PERSEUS
Workhorse
(WH)
study
is
a
prospective, randomized, controlled,
single-
blind, non-inferiority
trial
to
evaluate
the
safety and
efficacy
of
the
1 hg/mm
2
(loaded
drug/stent
surface area)
IONTM
Stent
in
the
treatment
of
de
novo
coronary lesions.
Subjects
with de
novo target
lesion length
<28mm and
target
vessel
diameter
>2.75mm
to
K4.Omm
were
considered
for
enrollment.
The
trial
employs
a
3:1
randomization
to
the
ION
or the
TAXUS
Express
Paclitaxel-Eluting
Stent
respectively.
The
primary
endpoint
is
the
rate
of
target
lesion failure
(TLF;
including
any
ischemia-
driven revascularization
of
the
target
lesion
[TLR],
myocardial
infarction
[MI;
Q-wave
and
non-Q-wave]
related
to
the
target vessel,
or
cardiac
death
related
to
the
target vessel)
at
12
months
post-index
procedure,
testing non-inferiority
of
the
ION
Stent relative
to
the
TAXUS
Express
Paclitaxel-Eluting
Stent
control.
In-segment percent diameter stenosis
at
9
months
post-index
procedure
as
measured
by
quantitative
coronary
angiography
(QCA)
is
the
secondary
endpoint.
Enrollment
of
1264
subjects
was
planned;
1262
(942
ION
Stent and
320
TAXUS
Express
Stent)
were
enrolled
and
randomized
at
90
sites.
A
total
of
330
subjects
were
randomly
assigned
to
protocol-mandated
9-month
angiographic follow-up
(angiographic subset).
The
protocol mandated
antiplatelet
therapy compliance
in
accordance
with
the
ACC/AHA/SCAI
Guidelines
for
PCI
2
.
The
study
is
now considered
complete with
regard
to
the 12-month
primary
endpoint.
Additional
follow-up
is
ongoing
to
5
years.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
19

PERSEUS
SV
The
PERSEUS
Small Vessel
(SV)
study
is
a
prospective,
single-arm,
superiority
trial
to
evaluate
the
safety
and
efficacy
of
the
1
tg/mm2
(loaded
drug/stent
surface
area)
2.25mm
and
2.5mm
IONTM
Stents
in
the
treatment
of
de
novo
coronary lesions
in
small
vessels.
Subjects
with
de
novo
target
lesion
length
<20mm
and target
vessel
diameter
>2.25mm
to
<2.75mm
in
a
native
coronary
artery
were
considered
for
enrollment.
The
trial
compares
the
ION
Stent
to
a
matched
bare metal
(Express
Stent)
historical
control
group
comprised
of
subjects
with reference vessel
diameter (RVD)
2.25 to
<2.75mm
and
lesion
length
20mm
from
the
TAXUS
V
trial.
The
TAXUS
V
trial
is
more
fully
described
in
the
SSED
for
P030025/SO28
(see
http://www.accessdata.fda.gov/cdrh
docs/pdf3/P030025SO28b.pdf).
All
subjects
in
PERSEUS
SV
were
required
to
undergo
a
9-month
angiographic
assessment.
The primary
endpoint
is
in-stent
late
loss
by
quantitative
coronary
angiography
(QCA)
on
9-
month
follow-up
(ION
Stent
compared
to
bare
metal
Express
Stent)
and
the
secondary
endpoint
is
TLF
at
12
months
(ION
Stent
compared
to
a
performance
goal
based
on
TAXUS
Express
Stent results from
the
TAXUS
IV
and
TAXUS
V
trials).
A
total
of
224
subjects
were
enrolled
at
28
sites. The
control
group
consisted
of
125
matched
bare
metal
Express
Stent
subjects from
the
TAXUS V
trial,
including
108
with
9-
month
QCA
follow-up.
The
protocol
mandated antiplatelet therapy compliance
in
accordance
with
the
ACC/AHA/SCAI
Guidelines
for
PCI
2
.
The
study
is
now considered complete with
regard
to
the
primary endpoint.
Additional
follow-up
is
ongoing
to
5
years.
Table
11:
Compari
on
of
PERSEUS
Clinical Studies
PERSEUS
Workhorse
PERSEUS
Small
Vessel
Purpose
Evaluation
of
safety
and
Evaluation
of
safety
and
effectiveness
in
workhorse
lesions
effectiveness
in
small
vessel
lesions
Prospective,
multicenter, Prospective,
multicenter,
single-
Study
Design
randomized, single-blind,
non-
arm,
superiority
to BMS
inferiority
to
PES
Primary
Endpoint
12-month TLF
9-month in-stent
late
loss
Total:
1264
planned;
1262
Total:
224
Number
of
Subjects enrolled
and
randomized
ION
Stent: 224
(ITT)
ION
Stent:
922
TAXUS
Express
stent:
320
Polymer
Slow
Release
Translute
m
Polymer
PTx
Dose
Density
1
gg/mm
Lesion
Criteria:
>2.75mm
to
<4.00mm
>2.25mm
to
<2.75mm
Vessel Diameter
(by
visual
estimate)
Lesion
Criteria:
Lesion
Length
(by
<28mm
<20mm
visual
estimate)
Number
of
stents
Single
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
20

Stent
Matrix
2.75-4.0mm
diameter
2.25-2.50mm
diameter
8,
12,
16,
20,
24,
28,
32mm
length
8,
12,
16,
20,
24mm
length
Post-Procedure
Clopidogrel
or
ticlopidine:
at
least
6
months,
ideally
for
12
months
in
Antiplatelet
subjects
who
are
not
at
high risk
for
bleeding.
Therapy
ASA:
indefinitely
Clinical:
30
day,
9
month,
1
year,
Clinical:
30
day,
9
month,
1
18
month,
annually
2-5
years
year,
18
month,
annually
2-5
Angiographic
(330
subject
subset):
years
9
month
Angiographic
(all):
9
month
Abbreviations:
ASA=aspirin;
BMS=bare
metal
stent;
ITT=intent-to-treat;
PES=paclitaxel-
eluting
stent;
PTx=paclitaxel;
TLF-target
lesion failure
POTENTIAL
ADVERSE
EFFECTS
OF
THE
DEVICE
ON
HEALTH
Below
is
a
list
of
potential
adverse
events
(in
alphabetical
order) which
may
be
associated
with
the use
of
a
coronary stent
in
native
coronary
arteries
including
but
are
not
limited
to:
*
Abrupt
stent
closure
*
Acute
myocardial
infarction
*
Allergic
reaction
to
anti-coagulant
and/or
antiplatelet
therapy,
contrast medium,
or
stent
materials
*
Angina
*
Arrhythmias,
including
ventricular
fibrillation
and
ventricular tachycardia
*
Arteriovenous
fistula
*
Cardiac
tamponade
*
Cardiogenic
shock/pulmonary
edema
*
Coronary
aneurysm
*
Death
*
Dissection
*
Emboli,
distal
(air,
tissue
or
thrombotic
material
or
material
from
devices(s)
used
in
the
procedure)
*
Heart
failure
*
Hematoma
*
Hemorrhage,
requiring transfusion
*
Hypotension/hypertension
*
Infection,
local
or
systemic
*
Ischemia,
myocardial
*
Pain,
access
site
*
Perforation
or
rupture
of
coronary artery
*
Pericardial
effusion
*
Pseudoaneurysm,
femoral
*
Renal
failure
*
Respiratory
failure
*
Restenosis
of
stented
segment
*
Stent
embolization
or
migration
*
Stent
thrombosis/occlusion
*
Stroke/cerebrovascular
accident
/TIA
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
21

*
Total
occlusion
of
coronary
artery
*
Vessel
spasm
*
Vessel
trauma
requiring
surgical
repair
or
reintervention
Potential
adverse
events
not
captured
above,
that
may
be
unique
to
the
paclitaxel
drug
coating:
*
Allergic/immunologic
reaction
to
drug
(paclitaxel
or structurally-related
compounds)
or
the
polymer
stent
coating
(or
its
individual
components)
*
Alopecia
*
Anemia
*
Blood
product
transfusion
*
Gastrointestinal
symptoms
*
Hematologic
dyscrasia
(including
leukopenia,
neutropenia,
thrombocytopenia)
*
Hepatic
enzyme
changes
*
Histologic changes
in
vessel
wall,
including
inflammation,
cellular
damage
or
necrosis
*
Myalgia/arthralgia
*
Peripheral
neuropathy
For
the
specific adverse
events
that
occurred
in
the clinical studies,
please
see
Section
IX,
Tables
13
and
20.
A.
PERSEUS
WH
Clinical
Trial
Primary
Objective:
The
primary
objective
of
the
PERSEUS
WH study
was
to
evaluate
the
safety
and
efficacy
of
the
IONM Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent
System
for the
treatment
of
de
novo
atherosclerotic lesions
of
up
to 28mm
in
length
(by
visual
estimate)
in
native
coronary
arteries
of
2.75mm
to
4.0mm
diameter
(by
visual
estimate)
compared
to
TAXUS
Express
Stent control.
Design:
PERSEUS
WH
is
a
prospective, randomized, controlled,
single-blind,
non-
inferiority trial
which employs
a
3:1
randomization
to
the
ION
or
the
TAXUS
Express
Paclitaxel-Eluting
Stent
respectively.
Eligible subjects
were
those
18
years
old with
documented
stable
angina
pectoris, unstable
angina pectoris,
or
documented
silent
ischemia
and
left
ventricular ejection
fraction (LVEF)
30%.
De
novo
target lesions
in
a
native coronary
artery
'with
diameter
stenosis
>50%,
reference
vessel diameter
>2.75mm
to
<4.0mm,
and
cumulative
lesion
length
<28mm
coverable
by
a single
study
stent
were
eligible.
Multiple
stenting
was
allowed
for
bail-out
only.
The
protocol
mandated
antiplatelet therapy compliance
in
accordance
with
the
ACC/AHA/SCAI
Guidelines
for
PCI
2
.
While subjects
in
this
trial
could
be
blinded,
the
operators
could
not,
as
the
identity
of
the
stent could
be
determined
by
the
operator on
the
basis
of
a
visual
comparison
of
stent
design prior
to
implantation.
Use
of
a
Clinical
Events
Committee
and
an
angiographic
core
lab
minimized
potential
bias
introduced
from
the
inability
to
completely
blind
all
study
participants.
Enrollment
of
1264
subjects was
planned.
A
total
of
1262
(942
ION
Stent and
320
TAXUS
Express
Stent)
were
enrolled
and
randomized
at
90
centers.
Of
the
1262
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
22

subjects
included
in
the
intent-to-treat
analysis
set,
a
total
of
1235
subjects
(922
ION
Stent
and
313
TAXUS
Express
Stent)
were
evaluable
for
the
12-month
primary
endpoint.
A
total
of
330
subjects
(256
ION,
74
TAXUS
Express)
were
randomly
assigned
to
protocol-mandated
9-month
angiographic
follow-up
(angiographic
subset).
Angiographic
assessments
were
performed
for
the
area
of
the
vessel
within
the
stent
margins
(in-stent)
and
the
areas
immediately
5
mm
proximal
and
distal
from the stent
margins
(analysis
segment).
An
angiographic
core
lab
was
utilized
for
analysis
of
angiography
data.
A
Clinical
Events
Committee
(CEC)
served
as
a
multidisciplinary
expert
group
responsible
for
the
independent
and
ongoing
adjudication
of
prespecified
clinical events,
including
all
reported deaths, myocardial
infarctions
(MI),
target vessel
revascularizations
(TVR),
and
stent
thromboses
(ST),
as
defined
by
the
clinical
protocol.
A
Data
Monitoring Committee
(DMC)
of
independent
experts
in
cardiology,
cardiovascular
interventional therapy,
and
biostatistics
worked
to
ensure
patient
safety
by
evaluating
accumulating
data
from
the
PERSEUS
Clinical
Program.
1.
Clinical
Inclusion
and
Exclusion
Criteria
Enrollment
in
the
PERSEUS
WH
study
was
limited
to
subjects
who
met
the
following
inclusion
criteria:
*
Subject
is
>
18
years
old
*
Eligible
for
percutaneous
coronary
intervention
(PCI)
*
Documented
stable
angina
pectoris,
or
documented
silent
ischemia
*
Acceptable candidate
for
coronary
artery
bypass
grafting
(CABG)
*
Left
ventricular
ejection
fraction
(LVEF)
is
>
30%
*
Subject
(or
legal
guardian)
understands
the
study
requirements
and
the
treatment
procedures
and
provides
written
Informed
Consent
before
any
study-specific tests
or
procedures
are
performed
*
Subject
willing
to
comply with
all
specified
follow-up evaluations
ANGIOGRAPHIC INCLUSION
(by
visual
estimate)
*
Target lesion
located
in
native coronary artery
*
Target
lesion
must
be
de
novo
*
Target lesion
diameter stenosis
>50%
*
Reference vessel
diameter
(RVD):
>2.75mm
to
<4.0mm
*
Cumulative
target
lesion
length (area
to
be
treated
must
be
completely
coverable
by
one
study
stent):
28mm
*
Target
lesion
is
successfully
pre-dilated.
Subjects
are
enrolled
only
after
successful
balloon catheter
pre-dilation
of
the
target
lesion
*
One
non-target
lesion
may
be
treated
in
a
non-target
vessel
*
Non-target
lesion
in
non-target
vessel
must
be
treated
with
a
commercially
available
TAXUS
stent
if
use
of
drug-eluting
stent
required
*
Treatment
of
a
non-target
lesion
(if
performed)
must
be
deemed
a
clinical
angiographic
success,
without
requiring
use
of
unplanned
additional stent(s)
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
23

*
Treatment
must
be
completed prior
to
treatment
of
target
lesion
Subjects
were
not
permitted
to
enroll
in
the
PERSEUS
WH
study
if
they met
any
of
the
following
exclusion
criteria:
*
Contraindication
to
ASA,
or
to
both
clopidogrel
and
ticlopidine
*
Known
hypersensitivity
to
paclitaxel
*
Known
allergy
to
stainless
steel
*
Known
allergy
to
platinum
*
Previous
treatment
of
the
target
vessel
with
any
anti-restenotic
drug-
coated
or
drug-eluting
coronary
stent
*
Previous treatment
of
the
target
vessel
with
a
bare metal
stent
(BMS)
within
9
months
of
the
index
procedure
*
Previous
treatment
of
any
non-target
vessel with
any
anti-restenotic
drug-
coated
or
drug-eluting
coronary
stent
within
9
months
of
the
index
procedure
*
Previous
treatment
with
intravascular
brachytherapy
in
the
target
vessel
*
Planned
PCI
or
CABG
post-index
procedure
*
Planned
or
actual
target vessel
treatment
with
an
unapproved
device,
directional
or
rotational coronary
atherectomy,
laser,
cutting
balloon
or
transluminal extraction
catheter
immediately prior
to
stent
placement
*
Myocardial
infarction
(MI)
within
72
hours
prior
to
the
index
procedure
as
defined per protocol
definition
(see
Section
4.2)
*
Cerebrovascular
accident
(CVA)
within
the
past
6
months
*
Cardiogenic
shock
characterized
by
systolic
pressure
<80mm
Hg
and/or
central
filling pressure
>20mm
Hg,
or cardiac
index
<1.8
liters/minute/m
2
or
intra-aortic
balloon pump
or
intravenous inotropes
are
needed
to
maintain
a
systolic
pressure
>
80mm
Hg
and
a
cardiac index
>1.8
liters/minute/m
2
*
Acute
or
chronic
renal
dysfunction
(creatinine
>
2.0
mg/dl
or
177
jtmol/l)
*
Any
prior
true anaphylactic reaction
to
contrast
agents;
defined
as
known
anaphylactoid
or
other
non-anaphylactic
allergic
reactions
to
contrast agents
that
cannot
be
adequately pre-medicated prior
to
the index
procedure
*
Leukopenia
(leukocyte count
<3.5 x
109/liter)
*
Thrombocytopenia
(platelet
count
<100,000/mm
3
)
*
Thrombocytosis
(>750,000/mm3)
*
Active peptic ulcer
or
active
gastrointestinal
(GI)
bleeding
*
Current
treatment,
or
past
treatment
within
12
months
of
the
index
procedure,
with
paclitaxel
or
other
chemotherapeutic
agent(s)
*
Anticipated treatment
with
paclitaxel
or oral
raparmycin
during
any
period
in
the
9
months
after
the
index
procedure
*
Male
or
female
with
known
intention
to
procreate within
9
months
after
the
index
procedure
*
Positive pregnancy
test within
7
days
before
the
index procedure,
or lactating
*
Life
expectancy
of
less
than
24
months
due
to
other
medical
conditions
*
Co-morbid
condition(s)
that
could
limit
the
subject's
ability
to
comply
with
study
follow-up
requirements
or
impact
the
scientific
integrity
of
the
study
*
Currently
participating
in
another investigational
drug
or
device
study
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
24

ANGIOGRAPHIC
EXCLUSION
(by
visual
estimate)
"
Target
lesion
in
left
main
artery,
whether
protected
or
unprotected
*
Target
lesion
is
a
chronic
total
occlusion
(TIMI
flow
<1)
*
Target
lesion
is
restenotic
*
Target
lesion
is
located
in
a
saphenous
vein
graft or
mammary
artery
graft
*
Target
lesion
is
accessed
via
saphenous
vein
graft
or
mammary
artery
graft
*
Target
lesion
is
<5mm
from
bare
metal
stent
(BMS)
*
Target
lesion
is
<5mm
from
ostium
*
Target
lesion
is
<5mm
from
a
side
branch vessel
2.0mm
in
diameter
(Exceptions:
subject
may
be
enrolled
if
side
branch
is
100%
occluded
or
if
side
branch
is
protected
with
a
patent
graft)
*
Untreated
lesions
with
>50%
diameter
stenosis
or
thought
to
impair
flow
remaining
in
target vessel
at
a
location
with
2.0mm
RVD
*
Target
lesion
and/or target
vessel
proximal
to
the
target
lesion
is
moderately or
severely
calcified
*
Target lesion
and/or
target
vessel
proximal
to
the
target
lesion
is
severely
tortuous
*
Target lesion
is
located
within
or
distal
to
a
>600
bend
in
the
vessel
*
Target
lesion
with
angiographic
presence
of
probable
or
definite
thrombus
*
Unprotected
left
main
coronary
artery
disease
*
Protected
left
main
coronary
artery
disease
with
target
lesion
in
LAD
or
LCX
(subject
may
be
enrolled
if
only
lesion
is
target
lesion
in
RCA)
2.
Follow-up
Schedule
All
subjects
were
scheduled
to
return
for
follow-up examinations
at
30
days,
9,
12
and
18
months,
and
2,
3,
4
and
5
years
post
index
procedure.
Subjects
received
the
examinations
outlined
in
Table
12.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
25
6

ON 0
CD
K
JN
w
o
CD~
C)
0
-!
00
--
o-
-
CA, c~
0
a
<
00
0z
0
0-
<
0
>
CD9
0
CD
00
0
r,
D0
~0
-
nc
Eno
0<
a-
-t0
CD
~
0
V
--
IA
o
co
P
-0 0
a
0 S-
~
~
aD
0
oo
'o
a
00 x
a,
0
'=
0 r-
= .= 0j( F .
03~
-Da
C
-0
0.
-~
00-
CD
0
CDS
w
co
0
o
a
-
0
C
0
)0
W0 -
o
0
-
:a
N
(a.
0
oD
o
0-
n
o0a
pq
a1ao
-
<
0-
0.
o D CD
00
N=
CD
0wC
0 >
0-
0
a

After
the
12-month
follow-up,
the
study
population
was
reduced
to
a
pre-specified
cohort
(Safety
Population),
which
consists
of
all
subjects who received
a
study
stent
(ION
Stent
or
TAXUS
Express
Stent).
The
study
is
now
considered complete
with
regard
to
the
12-month
primary
endpoint.
The key
timepoints
are
shown
below
in
the
tables
summarizing
safety
and
effectiveness.
3.
Clinical
Endpoints
The
primary
endpoint
is
the rate
of
target
lesion
failure (TLF;
including
any
ischemia-driven
revascularization
of
the
target
lesion
[TLR],
myocardial
infarction
[MI;
Q-wave and
non-Q-wave]
related
to
the target
vessel,
or
cardiac
death
related
to
the
target
vessel)
at
12
months post-index procedure, testing non-inferiority
of
the
ION
Stent relative to
the
TAXUS
Express
Paclitaxel-Eluting
Stent control.
In-
segment percent
diameter
stenosis
at
9
months
post-index procedure
as
measured
by
quantitative
coronary
angiography
(QCA)
is
the
secondary
endpoint.
Both
the
primary
and
secondary
endpoints
were
analyzed
under
a
Bayesian
framework.
Bayesian analyses
can be
interpreted
in
a
more intuitive
way
than
conventional
frequentist
analyses
through
the
posterior
distributions
they
produce.
These
posterior
distributions
give
the
probability
that
a
parameter
of
interest
(e.g.
the
difference
in
the
rate
of
TLF
across
treatment
groups)
lies
within
a
certain
range,
given the
data
observed.
Therefore,
Bayesian
methods
can
provide
a
posterior
probability
that
the
non-inferiority
hypothesis
is
true
given
the
data
observed,
whereas
the
frequentist
P
value
provides
the
probability
of
observing
data
as
or
more
extreme
than
that
observed
assuming
the
non-inferiority
hypothesis
is
false.
Al.
Accountability
of
PMA
Cohort
At
the
time
of
database lock,
of
1262
subjects enrolled
in
PMA
study,
97.86%
(1235)
subjects
are
available
for
analysis
at
the
completion
of
the
study,
the
12-
month
primary
endpoint.
Subject
Disposition
for
PERSEUS
WH
Study,
ITT,
All
Subjects
(N=
262)
TAXUS
ION
Total
Express
(N=942)
(N=1262)
(N=320)
All Subjects
Treated
-
1264
Treated,
Not
Randomized
-
-
2 a
Intent-to-Treat
(ITT)
Analysis
Set
320
942
1262
Death
<395
days
with
no
12-month
Clinical
Follow-
2
6
8
up
Performed
Eligible
for
12-month
Clinical
Follow-up
318
936
1254
12-month
Clinical
Follow-Up
Visit
Performed
97.8%
97.6%
97.7%
(311/318)
(914/936)
(1225/1254)
Office
Visit
41
136
177
Telephone
Contact
270
776
1046
No
12-month
Clinical Follow-up
Visit
Performed
7
22
29
Premature
Discontinuation
3
9
12
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
27
4*

Subject
Disposition
for
PERSEUS
WH
Study,
ITT,
All
Subjects
(N=1262)
TAXUS
ION
Total
Express
(N=942)
(N=1262)
(N=320)
Withdrew
Consent
0
4 4
Lost
to
Follow-up
1 2
3
Other
2 3
5 d
Death
>395
days
0
0 0
Missed
12-month
Follow-up
Visit
4
13
17
With
Later
Follow-up
Visit
Performed
1 3 4
No
Later
Follow-up
Visit
Performed
3
10
13
12-month
Clinical Follow-Up
or
Death'
97.8%
97.7%
97.7%
(313/320)
(920/942)
(1233/1262)
12-month
Clinical
Follow-Up
Subject
Accountability
97.2%
97.0%
97.1%
(311/320)
(914/942)
(1225/1262)
Numbers
are
%
(Count/Sample
Size)
or
Count.
a
Two
subjects
were
treated
without
randomization
and
therefore
are
not
included
in
the
analysis.
b
Subjects
who
died
prior
to
completion
of
follow-up
window
and
prior
to
completing
a
12-month
clinical
follow-up visit
are
considered
censored
and
are
excluded
from
calculation
of
proportion
of
subjects
who
completed
clinical
follow-up
visit.
c
Based
on
subjects
eligible
for
12-month
clinical
follow-up (excludes
deaths
within
395
days).
d
Four
subjects
withdrawn
at
the
investigator's
discretion
and
one
coded
as
a
"discontinuation"
(all
for
lack
of
follow-up)
Includes
subjects
who
have
died
in
both
the
numerator
and
the
denominator;
based
on
intent-to-treat
analysis
set.
f
Based
on
intent-to-treat
analysis
set.
Abbreviations:
ITT:
intent-to-treat;
TLF:
target
lesion
failure.
A2.
Study
Population
Demographics
and
Baseline
Parameters
Subjects
were
well-matched
for
baseline
demographics.
Age
was
slightly
lower
in
the
IONTM
Stent group
compared
to
the
TAXUS
Express
Stent control
(62±9.6
versus
63±9.5).
Baseline
lesion
characteristics:
Reference
vessel
diameter
was
2.78±0.48mm
and
2.75+0.47mm
in
the
ION
Stent
and
TAXUS
Express
Stent groups,
respectively,
with
baseline
lesion
length
of
14.2±6.1mm
and
14.1±5.8mm,
respectively.
Baseline
Demographic
and
Clinical
Characteristics,
ITT,
All
Subjects,
PERSEUS
WH
(N=1262)
TAXUS
Express
ION
(N=320) (N=942)
Male
68.8%
(220/320)
70.8%
(667/942)
Age
(yr)
63.5±9.5
(320)
62.2±9.6
(942)
(41.0,
82.0)
(33.0,
84.0)
Ethnicity
and
Race
Hispanic
or
Latino
1.9%
(6/320)
1.1%
(10/942)
Caucasian
92.5%
(296/320)
92.6%
(872/942)
Asian
1.9%
(6/320)
1.6%
(15/942)
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
28

Baseline
Demographic
and
Clinical
Characteristics,
ITT,
All
Subjects,
PERSEUS
WH
(N=1262)
Parameter
TAXUS
Express
ION
(N=320) (N=942)
Black,
of
African
heritage
4.1%
(13/320)
3.9%
(37/942)
Native
Hawaiian
or other
Pacific
Islander
0.3%
(1/320)
0.4%
(4/942)
American
Indian
or
0.0%
(0/320)
0.3%
(3/942)
Alaska
native
Other
0.0%
(0/320) 0.4%
(4/942)
Cardiac
History
Previous
PCI
25.2% (80/318) 23.0%
(216/941)
Previous
CABG
6.3%
(20/319)
4.5%
(42/942)
Previous
MI
19.0%
(60/316) 20.9%
(195/935)
CHF
7.5%
(24/318)
6.0%
(56/937)
Stable
Angina
66.6%
(213/320)
66.8% (629/941)
Unstable
Angina
21.3%
(68/320)
20.7%
(195/941)
Silent
Ischemia
12.2%
(39/320)
12.4%
(117/941)
Ejection
Fraction
(%)
57.8±9.8
(317)
58.0±9.3
(939)
(30.0,
98.0)
(30.0,
93.0)
Cardiac
Risk
Factors
Smoking,
Ever
69.5%
(216/311)
66.7%
(611/916)
Current
23.5% (73/311)
24.3%
(223/916)
Previous
46.0%
(143/311)
42.4%
(388/916)
Medically
Treated
Diabetes
25.0%
(80/320)
24.6%
(232/942)
Insulin
Requiring
7.5%
(24/320)
7.3%
(69/942)
Non-insulin
Requiring
17.5%
(56/320)
17.3%
(163/942)
Hyperlipidemia
Req.
76.4%
(243/318)
76.3%
(717/940)
Medication
Hypertension
Req.
Medication
80.3%
(256/319)
75.3%
(709/941)
Family
History
of
CAD
66.4%
(196/295)
67.0%
(589/879)
Comorbidities
History
of
PVD
11.3%
(36/319)
10.6%
(99/938)
History
of
TIA
or
CVA
7.5%
(24/320)
5.7%
(54/941)
History
of
TIA
4.4%
(14/319)
3.2%
(30/940)
History
of
CVA
3.8%
(12/318)
3.2%
(30/940)
Renal
Disease
3.4% (11/320)
4.8%
(45/940)
History
of
GI
Bleeding
2.2% (7/319)
1.0%
(9/939)
Numbers
are
%
(Count/Sample
Size)
or
Mean±SD
(N)
(Min, Max).
Abbreviations:
CABG:
coronary
artery
bypass
graft;
CAD:
coronary
artery
disease;
CHF:
congestive
heart failure;
CI:
confidence
interval;
CVA:
cerebrovascular
accident;
GI:
gastrointestinal;
MI:
myocardial
infarction;
PCI:
percutaneous
coronary
intervention;
PVD:
peripheral
vascular
disease;
TIA:
transient
ischemic
attack.
Baseline
Lesion
Characteristics
by
QCA,
ITT,
All
Subjects,
PERSEUS
WH
(N=1262)
Lesion
Characteristic
TAXUS
Express
ION
(N=320)
(N=942)
Target Lesion
Vessel
LAD
43.8%
(140/320)
43.1%
(406/942)
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
29

LCX
21.6%
(69/320)
23.0%
(217/942)
RCA
34.7%
(111/320)
33.9%
(319/942)
Lesion
Location
Ostial
2.5%
(8/320)
2.7%
(25/942)
Proximal 40.0%
(128/320)
39.1%
(368/942)
Mid
48.1%
(154/320)
51.5%
(485/942)
Distal
9.4%
(30/320)
6.8%
(64/942)
Reference
Vessel
2.8±0.5
(320)
2.8±0.5
(942)
Diameter
(RVD,
mm)
(1.6,
4.6)
(1.4,
4.5)
Minimum
Lumen
0.8±0.4
(320)
0.8±0.3
(942)
Diameter
(MLD,mm)
(0.0,
2.1)
(0.0, 2.4)
Percent Diameter
Stenosis
71.7±10.9
(320)
72.1±10.9
(942)
(%
DS)
(40.0,
100.0)
(40.3,
100.0)
Lesion
Length
(mm)
14.1±5.8
(320)
14.2±6.1
(942)
(4.2,
40.9)
(3.2;
40.1)
Eccentric Lesion
50.3%
(161/320)
51.7%
(487/942)
Bend
(degrees)
>
45
6.9%
(22/320)
6.5%
(61/942)
>
90
0.3%
(1/320)
0.2%
(2/942)
Tortuosity,
Any
4.4%
(14/320)
6.3%
(59/942)
Moderate
4.1%
(13/320)
5.7%
(54/942)
Severe
0.3%
(1/320)
0.5%
(5/942)
Calcification,
Any
26.9%
(86/320) 24.2%
(228/942)
Moderate
22.2%
(71/320)
17.8%
(168/942)
Severe
4.7%
(15/320)
6.4%
(60/942)
Total
Occlusion
0.6%
(2/320)
0.6%
(6/942)
Bifurcation
40.3%
(129/320)
36.4%
(343/942)
25.7±28.8
(129)
24.4±28.1
(342)
(0.0,
95.0)
(0.0,
100.0)
Modified
ACC/AHA
A
8.8%
(28/320)
8.4%
(79/942)
BI
26.6% (85/320)
24.7%
(233/942)
B2
40.6%
(130/320)
40.8%
(384/942)
C
24.1%
(77/320)
26.1%
(246/942)
Pre-Procedure
TIMI
Flow
0
0.6%
(2/320)
0.2%
(2/942)
1
0.0%
(0/320)
0.4%
(4/942)
2
1.3%
(4/320)
3.6%
(34/942)
3
98.1%
(314/320)
95.8%
(902/942)
Numbers
are
%
(Count/Sample
Size)
or
Mean±SD
(N)
(Min,
Max).
Abbreviations:
ACC/AHA:
American
College
of
Cardiology/American
Heart
Association;
CI:
confidence
interval;
DS:
diameter
stenosis;
LAD:
left
anterior
descending;
LCX: left
circumflex;
MLD:
minimum
lumen
diameter;
QCA:
quantitative
coronary
angiography;
RCA:
right
coronary
artery; RVD:
reference
vessel
diameter;
TIMI:
thrombolysis
in
myocardial infarction.
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
30

A3.
Safety
and
Effectiveness
Results
1.
Safety
Results
The
analysis
of
safety
was
based
on
the
ION
stent
cohort
of
942
subjects
available
for the 12-month
primary
endpoint
evaluation.
Principal adverse
events
for
this
study
are
presented
in
Table
13
below.
Table
13:
Principal
Adverse
Events
PERSEUS
Workhorse
to
1
Year
TAXUS
ION
Stent Express
(N=942)
Stenta
(N=320)
In-Hospital
MACE
1.9%
(18/942)
2.5%
(8/320)
30-Day
MACE
2.2%
(21/939)
3.1%
(10/319)
9-Month
MACE
5.6%
(52/932)
6.3%
(20/317)
Cardiac
Death
0.3%
(3/932)
0.3%
(1/317)
MI
2.0%
(19/932)
2.8%
(9/317)
Q-Wave
MI
0.4%
(4/932)
0.0%
(0/317)
Non-Q-Wave
MI
1.6%
(15/932)
2.8%
(9/317)
TVR
4.0%
(37/932)
4.4%
(14/317)
TLR
2.6%
(24/932)
3.5%
(11/317)
Non-TLR
1.9%
(18/932)
1.3%
(4/317)
1-Year
MACE 7.4%
(68/922)
7.7%
(24/313)
Cardiac Death
0.5%
(5/922)
0.3%
(1/313)
MI
2.2%
(20/922)
2.9%
(9/313)
Q-Wave
MI
0.5%
(5/922)
0.0%
(0/313)
Non-Q-Wave
MI
1.6%
(15/922)
2.9% (9/313)
TVR
5.6%
(52/922)
5.8%
(18/313)
TLR
3.8%
(35/922)
4.5% (14/313)
Non-TLR
2.5%
(23/922)
1.96/(6/313)
1-Year
ARC
Stent
Thrombosis
Definite
or
Probable
0.4%
(4/918)
0.3%
(1/313)
Definite
0.3%
(3/918)
0.3%
(1/313)
Probable
0.1%
(1/918)
0.0% (0/313)
'DES
Control
An
angiographic
core
laboratory
review
of
all
available
angiograms
in
the
PERSEUS
Clinical
Trial
revealed
a
total
of
3
stent fractures
-
2
ION
stent fractures
(Type
33
fractures
that
were
seen
on angiograms
performed
at 286
and
259
days
post-stent
implantation)
and
1
Taxus Express
stent
fracture
(Type
43
fracture
noted
on
an
angiogram
performed
861
days
post-stent
implantation).
Only
the fracture
that
occurred
with
the
Taxus
Express stent
was
associated
with
a
major
adverse
cardiovascular
event
(a
TLR).
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
31

2.
Effectiveness
Results
The
analysis
of
effectiveness
was
based
on the
942
subjects
available
for
the
12-
month
primary
endpoint
evaluation.
Key
safety
and
effectiveness
outcomes
are
presented
in Tables
14
through
19.
Table
14:
PERSEUS
Workhorse
Clinical
Results
1-year
(ITT
population)
ION
Stent
TAXUS
Express
Stent'
(N=942)
(N=320)
EFFICACY
TVR,
Overall
5.6%
(52/922)
5.8%
(18/313)
TLR,
Overall
3.8%
(35/922)
4.5%
(14/313)
TLR,
PCI
3.6%
(33/922)
4.2%
(13/313)
TLR,
CABG
0.3%
(3/922)
0.6%
(2/313)
Non-TLR,
Overall
2.5%
(23/922)
1.9%
(6/313)
Non-TLR,
PCI
2.3%
(21/922)
1.6%
(5/313)
Non-TLR,
CABG
0.3%
(3/922)
1.0%
(3/313)
SAFETY
Total
Death
0.7%
(6/922)
0.6%
(2/314)
Cardiac
Death
or MI
2.5%
(23/922)
2.9%
(9/313)
Cardiac
Death
0.5%
(5/922)
0.3%
(1/313)
MI
2.2%
(20/922)
2.9%
(9/313)
Q-wave
MI
0.5%
(5/922)
0.0% (0/313)
Non-Q-Wave
MI
1.6%
(15/922)
2.9%
(9/313)
ARC
Stent Thrombosis
Definite
or
Probable
0.4%
(4/918)
0.3%
(1/313)
Definite
0.3%
(3/918)
0.3%
(1/313)
Probable
0.1%
(1/918)
0.0% (0/313)
a
DES
Control
b
Timing
of
non-Q-wave
Ml:
15/15 ION
events
and
8/9
TAXUS
Express events
occurred
peri-procedurally.
This
trial
was
not sized
to
determine
the
rate
of
low
frequency
events
with
a
pre-specified
precision.
Abbreviations:
ARC=Academic
Research
Consortium;
CABG=coronary
artery
bypass
graft;
DES=drug-eluting
stent;
MI=myocardial
infarction;
PCI=percutaneous
coronary
intervention;
TLR=target
lesion
revascularization;
TVR=target
vessel
revascularization.
Primary
Endpoint
(12-Month
TLF):
The
primary
endpoint
was
met:
There
is
a
99.96%
Bayesian
posterior
probability
that
ION
Stent
is
non-inferior
to
TAXUS
Express
Stent (given
the
data observed),
demonstrating non-inferiority
of
the
ION
Stent
versus
the
TAXUS
Express
Stent.
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness Data
Page
32

Table
15:
PERSEUS
Workhorse
Primary
Endpoint
TAXUS
Express
12-Month
ION
Stent
Stent
Difference
Posterior
Target
Lesion
Posterior Posterior
Posterior
b
Probabilit
95%
C
Failure
(TLF)
Mean
(SD)
Mean
(SD)
Mean
(SD)
A
y
of
NI
c
Per
Protocola
5.568%
6.38%
-0.570%
1.85%
4.1%
0.9996
(0.0076)
(0.0136)
(0.0155)
a
Primary
analysis for
assessing hypothesis
of
non-inferiority
and
study
success criterion.
For
per
protocol analyses, only
ITT
PERSEUS
Workhorse
trial
subjects
who
had
the
randomly
assigned
study
stent implanted
in
the
target
coronary
artery were included.
b
1-Sided
950Z
posterior
credible
interval,
based
off
the
95th
percentile
of
the
posterior
distribution.
Posterior probability
that
the
difference
in
the
rate
of
12-month
TLF between
ION
Stent and
TAXUS
Express
Stent
is
less
than
the
pre-specified
margin
of
4.1%,
given the data.
12-Month
TLF:
the
proportion
of
subjects
who
experience
a
TLF
up
to
365
days
post-procedure
out
of
the
population
that
have been
followed
for
at least
335
days or who
have
experienced
a
TLF
up
to
335
days
post-procedure.
Secondary
Endpoint
(9-Month
%DS):
The
secondary
endpoint
was
met:
There
is
a
99.70%
Bayesian posterior
probability
that
IONTM
Stent
is
non-inferior
to
TAXUS
Express
Stent (given
the data observed),
demonstrating
non-inferiority
of
the
ION
Stent versus
the
TAXUS
Express Stent.
Table
16:
PERSEUS
Workhorse
Secondary
Endpoint
TAXUS
Ln
(9-
Express
Month
In-
Stent
ION
Stent
Difference
Posterior
Segment
Posterior Posterior
Posterior
95%
CIb
Probabilit
%DS)
Mean
(SD)
Mean
(SD)
Mean
(SD)
A
y
of
NI
C
Per
3.117
3.087
-0.0294
01078
0.20
0.9970
Protocola
(0.0736)
(0.0374)
(0.08253)
1
1
a
Primary
analysis
for
assessing
hypothesis
of
non-inferiority.
For
per
protocol
analyses,
only
ITT
PERSEUS
Workhorse
trial
subjects who
had
the
randomly assigned
study stent implanted
in
the
target
coronary artery
were included.
b
1-Sided
95%
posterior
credible
interval,
based
off
the
95th
percentile
of
the
posterior
distribution.
Posterior
probability
that
the
difference
in
mean
In
(9-month
%DS)
between
ION
Stent
and
TAXUS
Express
Stent
is
less
than
the
pre-specified
margin
of
0.20, given
the
data.
The
secondary
endpoint
is
9-month
in-segment
%DS.
For
the
secondary
endpoint,
a
natural
log
(In)
transformation
was
used
to
improve
the
normality
of
the
distribution.
Analyses
are
performed
on
the
transformed
data.
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
33

Table
17:
PERSEUS
Workhorse
Angiographic
Results
TAXUS
Express
ION
Stent
Stentb
Angiographic
Outcomes
a(N=228)
(N=61)
MLD
(mm),
In-stent
Post-Procedure
2.68±0.39
(228)
2.54±0.36
(61)
9-Month
2.34±0.67
(228)
2.28±0.64
(61)
MLD
(mm),
Analysis
Segment
Post-Procedure
2.25±0.49
(228)
2.16±0.37
(61)
9-Month
2.08±0.63
(228)
2.00±0.56
(61)
Acute
Gain
(mm),
In-stent
1.93±0.41
(228)
1.83±0.40
(61)
Acute
Gain,
Analysis
Segment
1.51±0.48
(228)
1.45±0.40
(61)
(mm)
%
DS,
In-stent
Post-Procedure
4.10±10.13
(228)
5.64±8.05
(61)
9-Month
16.37±20.86
(228)
16.02±20.61
(61)
%
DS,
Analysis
Segment
Post-Procedure
20.21±9.71
(228)
19.87±7.57
(61)
9-Month
26.10±17.71
(228)
26.37±17.47
(61)
Late Loss,
In-stent
(mm)
0.34±0.55
(228)
0.26±0.52
(61)
Late Loss,
Analysis
Segment
(mm)
0.17±0.48
(228)
0.16±0.45
(61)
Binary
Restenosis
In-stent
restenosis
7.9%
(18/228)
6.6%
(4/61)
Analysis
segment
restenosis
8.8%
(20/228)
9.8%
(6/61)
a
Includes
all
subjects
in
the
angiographic
subset
with
paired lesion
data.
b
DES
Control
Abbreviations:
DES=drug-eluting
stent;
DS=diameter
stenosis;
MLD=minumum
lumen
diameter.
Table
18:
PERSEUS
Workhorse
Stent
Thrombosis
ION
Stent
TAXUS
Express
Stentc
(N=942)
Set
Intent-to-Treat
Population
(N=320)
Protocol
Defined
Stent
Thrombosis
a
Cumulative
through
1
year
0.4%
(4/918)
0.3%
(1/313)
Acute
ST
0.2%(2/942)
0.3%
(1/320)
(524
hrs)
Subacute
ST
0.0%
(0/939)
0.0%
(0/319)
(>24
hrs
and
!30
days)
Late
ST
(>30
ays
nd
2
moths)0.2%
(2/936)
0.0%
(0/3
17)
(>30
days
and:512
months)
i
ARC
Definite
&
Probable
Stent
Thrombosis
b
Cumulative
through
1
year
0.4%
(4/918)
0.3% (1/313)
Acute
ST
0.2%
(2/942)
0.3%
(1/320)
(S24
hrs)
Subacute
ST
(>24hrs
nd
30
dys)0.0%
(0/939)
0.0%
(0/3
19)
(>24
hrs
and:530
days)
Late
ST
(>30
ays
nd
<2
moths)0.2%
(2/936)
0.0%
(0/3
17)
(>30 days
and:512
months)
To
be
included
in
the
calculation
of
stent
thrombosis
(ST)
rate
for
a
given
interval,
a
patient
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
34

either
had
to have
a
stent
thrombosis
during
the
interval
(e.g.
31-365
days
inclusive)
or they
had
to
be
stent
thrombosis-free
during
the
interval
with
last
follow-up
on
or
after
the
first
day
of
the given interval (e.g.
31
days).
a
Per
protocol,
stent
thrombosis
is
defined
as
the
occurrence
of
any
of
the
following:
1.
Clinical
presentation
of
acute
coronary syndrome
with
angiographic
evidence
of
stent
thrombosis:
a)Angiographic
documentation
of
a
complete occlusion
(TIMI
flow
0
or
1)
of
a
previously
successfully treated
artery (TIMI
flow
2
to
3
immediately
after stent placement
and
diameter
stenosis
530%)
and/or
b)
Angiographic
documentation
of
a
flow-limiting thrombus
within
or
adjacent
to
a
previously
successfully
treated
lesion.
2.
Acute
MI
of
the
distribution
of
the
treated
vessel.
3.
Death within
the first
30
days
(without
other
obvious
cause)
is
considered
a
surrogate
for
stent
thrombosis
when
angiography
is
not available.
b
Academic
Research
Consortium
(ARC)
stent
thrombosis
is
defined
as
follows
4
:
1.
Definite
ST
is
considered
to
have
occurred
after
intracoronary
stenting
by
either angiographic
or
pathologic
confirmation
of
stent
thrombosis.
2.
Probable
ST
is
considered
to have
occurred
after intracoronary
stenting
in
the
following
cases:
a)
Any
unexplained
death
within
the
first
30
days
following
stent
implantation.
b)
Irrespective
of
the
time after
the index
procedure,
any
MI
which
is
related
to
documented
acute
ischemia
in
the
territory
of
the
implanted stent
without angiographic
confirmation
of
ST
and
in
the
absence
of
any
other obvious
cause.
DES
Control
Numbers
are
%
(Count/Sample
Size).
This
trial
was
not sized to
determine
the
rate
of
low
frequency
events
with
a
pre-specified
precision.
Figure
3:
PERSEUS
Workhorse
Cumulative
Rate of
Target
Lesion
Failure
to
12
Months,
Intent-to-Treat,
Event Rate
±
1.5
SE,
All
Subjects
(N=1262)
20
-
woo
TAXUS
Express
Stent
(N=320)
ION
Stent
(N=942)
g 15
N
10
6.0%
0
0
30
60
90 120
150
180
210 240
270 300 330
360
390
Days
Since
Index
Procedure
TAXUS
Express
Stent:
320
311
310
309 308
307 306 305
301
ION
Stent:
942
921
918 914 913 910
905
900
887
Event Rate Event
Free
ION
Stent
5.6%
94.4%
TAXUS
Express
Stent
DES
6.0%
94.0%
Control
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data Page
35
/
Figure
4:
PERSEUS
Workhorse
Cumulative
Frequency
Distribution
of
9-Month
In-
Segment
Percent
Diameter
Stenosis
by
QCA,
Intent-to-Treat,
All
Angiographic
Subset
Patients
(N=330)
100
-
80 -
m
ION
,A E I TAXULS
Stenit
Express
Stent
60
- ,
(N=228) (N=61)
- ----
- - ---
- -- -
- - --- -- -
- - - Median
20.57
20.81
a)
Minimum
3.31
7.56
40
-
Maximum
100
100
Mean
26.1
26.37
20
-
I
Standard
Deviation
17.71
17.47
Coefficient
of Var. 67.84% 66.25%
0*
0
20
40
60
80
100
Observed
Value
Results
in
subjects
with and
without
diabetes: Subjects
with diabetes
mellitus
represent
a
high-risk
group
for
adverse
events
following percutaneous
coronary
intervention. Table
19
shows
1-year
outcomes
in
subjects
with
and
without
medically
treated
diabetes (defined
as
treatment
with
oral
hypoglycemic agents
or
insulin
at
enrollment).
While
the
PERSEUS
WH
study
randomization
was
stratified
for
diabetic
status,
this trial
was
not
adequately
powered
to
study
safety
or
effectiveness
of
the
TAXUS
Express
Stent
versus
the
ION
Stent
in
subjects
with
or
without
diabetes
and was
not
designed
to
specifically support
an
approval
for use
in
diabetic subjects.
These
exploratory
analyses
suggest that
in
subjects
treated
with
the
ION
Stent,
1-year
TLR
rates
were
4.9%
in
diabetic
and
3.4%
in
non-diabetic
subjects.
Table
19:
PERSEUS
Workhorse
Clinical
Results
in
Subjects with
and
without
Medically
Treated
Diabetes
I-year
(ITT
Population)
ION
Stent
Subjects
With
ION
Stent
Subjects
Medically
Treated
Without
Medically
Treated
Diabetes
Diabetes
(N=232)
(N=710)
EFFICACY
TVR,
Overall
7.6%
(17/223)
5.0%
(35/699)
TLR,
Overall
4.9%
(11/223)
3.4%
(24/699)
TLR,
PCI
4.5%
(10/223)
3.3%
(23/699)
TLR,
0.4%
(1/223)
0.3%
(2/699)
CABG
Non-TLR,
3.6%
(8/223)
2.1%
(15/699)
Overall
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
36
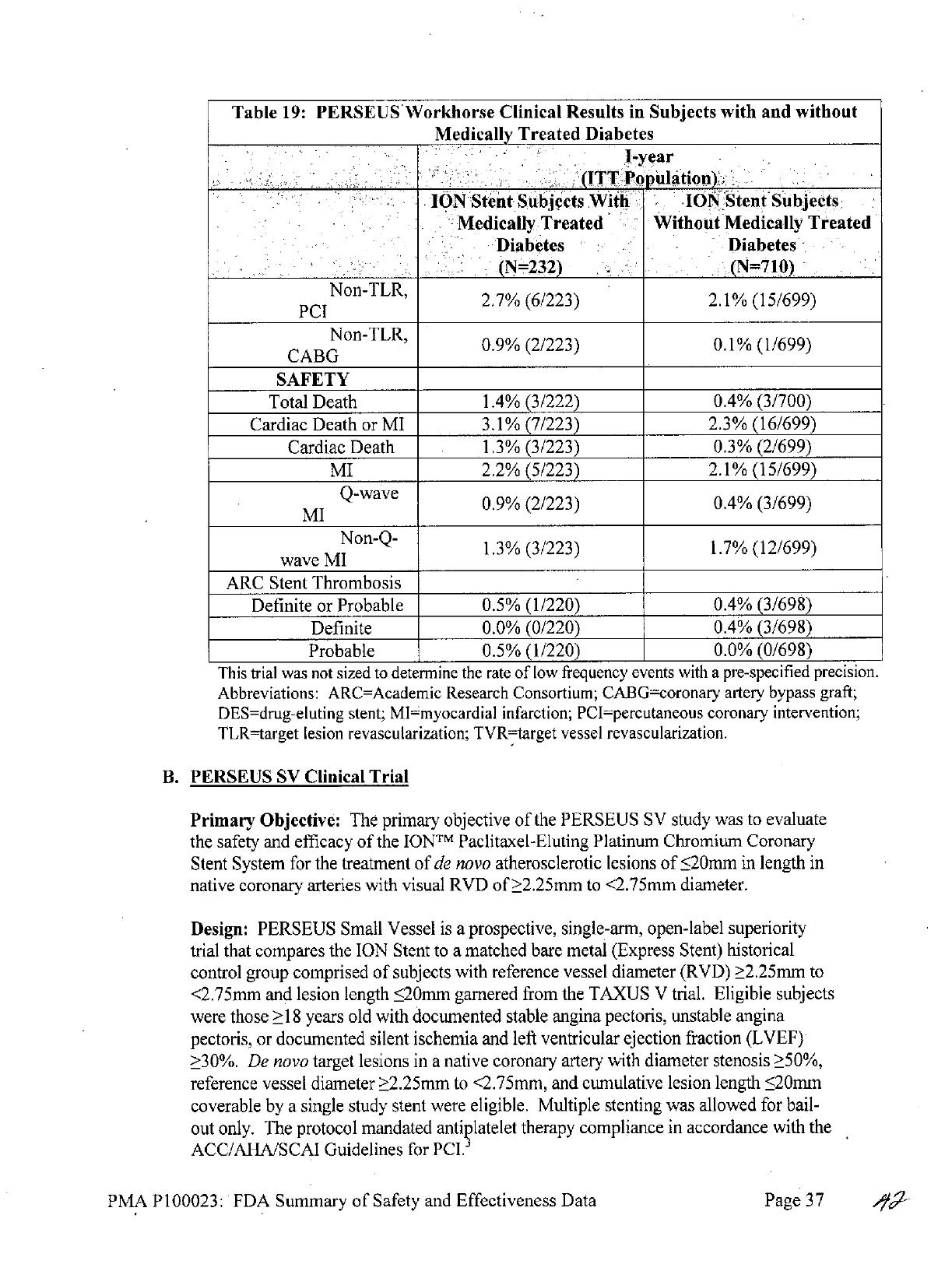
Table
19:
PERSEUS
Workhorse
Clinical
Results
in
Subjects
with
and without
Medically
Treated
Diabetes
I-year
(ITT
Population)
ION
Stent
Subjects
With
ION
Stent
Subjects
Medically
Treated
Without
Medically
Treated
Diabetes
Diabetes
(N=-232)
(N=7
10)
Non-TLR,
2.7%
(6/223)
2.1%
(15/699)
PCI
Non-TLR,
0.9%
(2/223)
0.1%
(1/699)
CABG
SAFETY
Total
Death
1.4%
(3/222)
0.4% (3/700)
Cardiac
Death
or
MI
3.1%
(7/223)
2.3%
(16/699)
Cardiac
Death
1.3%
(3/223)
0.3%
(2/699)
MI
2.2%
(5/223)
2.1%
(15/699)
MI
Q-wave
0.9%
(2/223)
0.4%
(3/699)
Non-Q-
1.3%
(3/223)
1.7%
(12/699)
wave
MI
ARC
Stent
Thrombosis
Definite
or
Probable
0.5%
(1/220)
0.4% (3/698)
Definite
0.0%
(0/220)
0.4% (3/698)
Probable
0.5%
(1/220)
0.0%
(0/698)
This
trial was not
sized
to
determine
the
rate
of
low
frequency
events with
a
pre-specified
precision.
Abbreviations:
ARC=Academic
Research
Consortium;
CABG=coronary
artery
bypass
graft;
DES=drug-eluting
stent;
MI~myocardial
infarction;
PCI=percutaneous
coronary
intervention;
TLR-target
lesion
revascularization;
TVR=target
vessel
revascularization.
B.
PERSEUS
SV
Clinical
Trial
Primary
Objective:
The
primary
objective
of
the
PERSEUS
SV
study
was
to
evaluate
the
safety
and
efficacy
of
the
ION
TM
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent
System
for the
treatment
of
de
novo
atherosclerotic
lesions
of
<20mm
in
length
in
native
coronary
arteries
with
visual
RVD
of
>2.25mm
to
<2.75mm
diameter.
Design:
PERSEUS
Small
Vessel
is
a
prospective,
single-arm,
open-label
superiority
trial
that compares
the
ION
Stent
to
a
matched
bare
metal
(Express
Stent)
historical
control
group
comprised
of
subjects
with
reference
vessel
diameter
(RVD)
>2.25mm
to
<2.75mm
and
lesion
length
<20mm
garnered
from
the
TAXUS
V
trial.
Eligible
subjects
were
those
>18
years
old
with
documented
stable
angina
pectoris,
unstable angina
pectoris,
or
documented
silent
ischemia
and
left
ventricular
ejection
fraction
(LVEF)
>30%.
De
novo
target
lesions
in
a
native
coronary
artery
with
diameter
stenosis
>50%,
reference
vessel
diameter
>2.25mm
to
<2.75mm,
and
cumulative
lesion
length
520mm
coverable
by
a
single
study
stent
were
eligible.
Multiple
stenting was
allowed
for
bail-
out
only.
The
protocol
mandated
antiqlatelet
therapy
compliance
in
accordance
with
the
ACC/AHA/SCAI
Guidelines
for
PCI.
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
37

Enrollment
of
224
subjects
was
planned.
A
total
of
224
subjects
were
enrolled
at
28
centers.
Of
the
224
subjects
included
in
the
intent-to-treat analysis
set, a
total
of
197
subjects
were
evaluable
for the
9-month
primary
endpoint
of
in-stent
late
loss.
The
control
group
consisted
of
125
matched
bare metal
Express
subjects
from
the
TAXUS
V
trial,
including
108
with 9-month
QCA
follow-up.
An
angiographic
core
lab
was
utilized
for
analysis
of
angiography
data.
A
Clinical
Events
Committee
(CEC)
served
as
a
multidisciplinary
expert
group
responsible
for
the
independent
and
ongoing adjudication
of
prespecified
clinical events,
including
all
reported deaths, myocardial
infarctions
(MI),
target
vessel
revascularizations
(TVR),
and
stent
thromboses
(ST),
as
defined
by
the
clinical
protocol.
A
Data
Monitoring
Committee
(DMC)
of
independent
experts
in
cardiology,
cardiovascular
interventional
therapy,
and
biostatistics
worked
to
ensure
patient
safety
by
evaluating
accumulating
data
from
the
PERSEUS
Clinical
Program.
1.
Clinical
Inclusion
and
Exclusion
Criteria
The
clinical
inclusion
and
exclusion
criterion
are
identical
to
those
described
in
Section
IX.A.
1.
with
the
following
exception:
ANGIOGRAPHIC
INCLUSION
(by
visual
estimate)
*
Reference
vessel
diameter
(RVD):
>2.25mm
to
<2.75mm
*
Cumulative
target lesion length
(area
to
be
treated
must
be
completely
coverable
by
one
study
stent):
<20mm
2.
Follow-up
Schedule
Follow-up
included
clinical
assessments
at
30
days,
9,
12
and
18
months,
and
2,
3,
4
and
5
years
post
index
procedure.
The
same
examination
schedule
as
outlined
in
Table
12,
above,
was utilized
for the
PERSEUS
SV
trial,
with
one
exception.
All
224
enrolled
subjects
were
required
to
undergo
protocol-
mandated
9-month
angiographic
follow-up.
Angiographic
assessments
were
performed
for
the
area
of
the
vessel
within
the
stent
margins
(in-stent)
and
the
areas
immediately
5
mm
proximal
and
distal
from
the
stent
margins
(analysis
segment).
After
the 12-month
follow-up,
the
study
population
was reduced
to
a pre-
specified
cohort
(Safety
Population),
which
consists
of
all
subjects
who
received
a
study
stent
(ION
Stent).
The
study
is
now considered
complete
with
regard
to
the
9-month
primary
and
12-month
secondary
endpoints.
3.
Clinical
Endpoints
The
primary
endpoint
is
in-stent
late
loss
by
QCA
on
9-month
follow-up
(ION
Stent
compared
to
bare
metal
Express
Stent)
and
the
secondary
endpoint
is
target
lesion
failure
(TLF;
including
any
ischemia-driven
revascularization
of
the
target
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
38
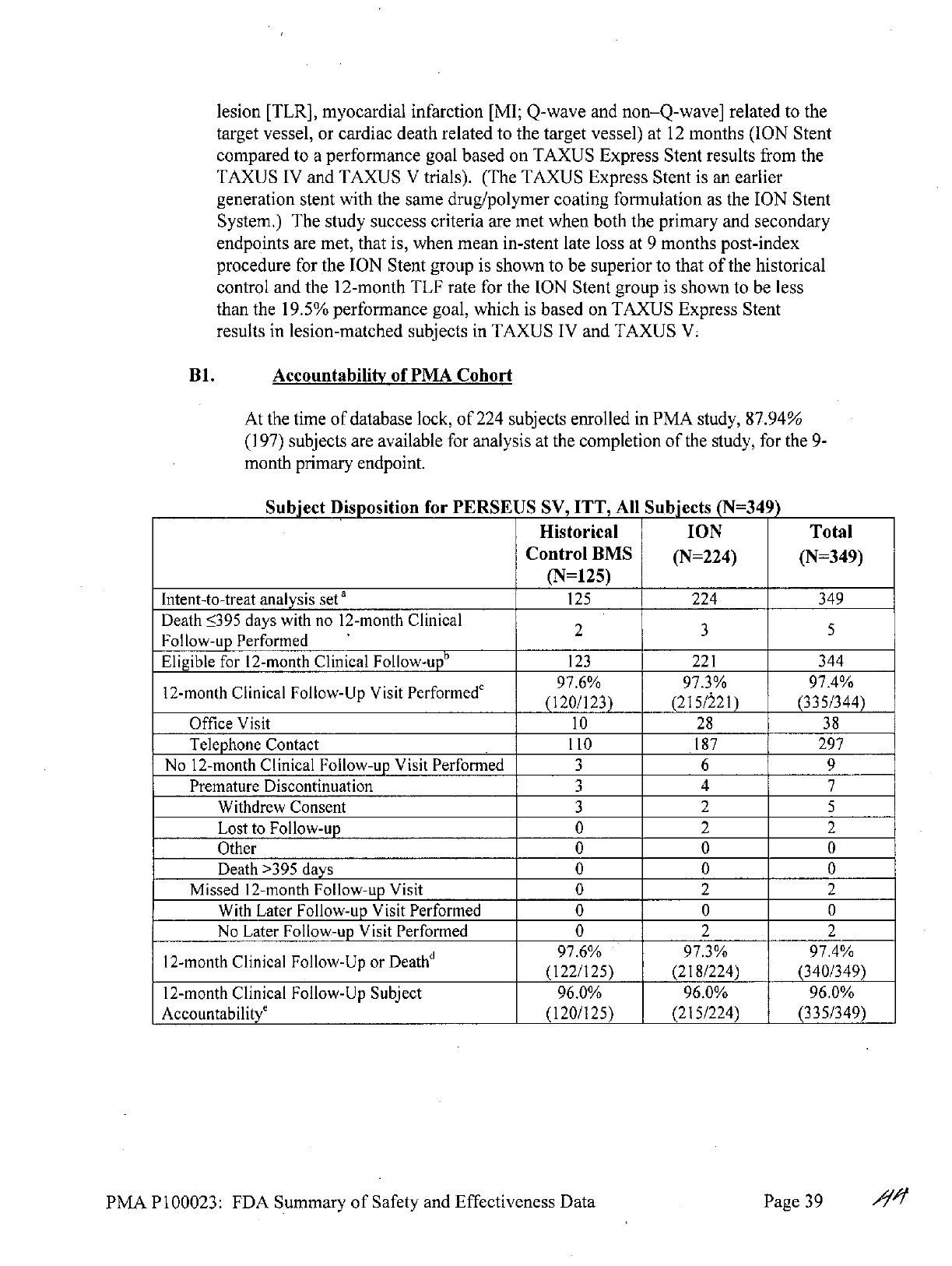
lesion
[TLR],
myocardial infarction
[MI;
Q-wave
and
non-Q-wave]
related
to
the
target vessel,
or
cardiac death related
to
the
target
vessel)
at
12
months
(ION
Stent
compared
to
a
performance
goal
based
on
TAXUS
Express
Stent
results
from
the
TAXUS IV
and
TAXUS
V
trials).
(The
TAXUS
Express
Stent
is
an
earlier
generation stent with
the same
drug/polymer
coating
formulation
as
the
ION
Stent
System.)
The
study
success criteria
are
met
when both
the
primary
and
secondary
endpoints
are
met, that
is,
when
mean
in-stent
late
loss
at
9
months post-index
procedure
for
the
ION
Stent
group
is
shown
to
be
superior
to
that
of
the
historical
control
and
the
12-month
TLF
rate
for the
ION
Stent
group
is
shown
to
be
less
than
the
19.5%
performance
goal,
which
is
based
on
TAXUS
Express
Stent
results
in
lesion-matched
subjects
in
TAXUS
IV
and
TAXUS
V.
B1.
Accountability
of
PMA
Cohort
At
the
time
of
database lock,
of
224
subjects
enrolled
in
PMA
study,
87.94%
(197)
subjects
are
available
for
analysis
at
the
completion
of
the
study,
for the
9-
month primary
endpoint.
Subject
Disposition
for
PERSEUS
SV,
ITT,
All
Subjects
(N=349)
Historical
ION
Total
Control
BMS
(N=224)
(N=349)
(N=125)
Intent-to-treat
analysis
set
a
125
224
349
Death
5395
days
with
no
12-month
Clinical
2
3
5
Follow-up Performed
Eligible
for
12-month
Clinical
Follow-upb
123
221
344
97.6%
97.3%
97.4%
12-month
Clinical
Follow-Up Visit
Performede
(2/2)25/13534
(120/123)
(215/
221)
(335/344)
Office
Visit
10
28
38
Telephone
Contact
110
187
297
No
12-month
Clinical
Follow-up
Visit
Performed
3 6 9
Premature
Discontinuation
3 4 7
Withdrew
Consent
3
2 5
Lost
to
Follow-up
0 2
2
Other
0 0
0
Death
>395
days
0 0
0
Missed
12-month
Follow-up Visit
0 2 2
With
Later
Follow-up
Visit
Performed
0 0
0
No Later
Follow-up
Visit
Performed
0 2
2
d
97.6%
97.3%
97.4%
12-month
Clinical
Follow-Up
or
Deathd(2/5)2124)3049
(122/125)
(218/224)
(340/349)
12-month
Clinical
Follow-Up
Subject
96.0%
96.0%
96.0%
Accountabilitye
(120/125)
(215/224)
(335/349)
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
39

Numbers
are
%
(Count/Sample
Size)
or
Count.
a.
Intent-to-treat
is
the
primary
analysis
set
for
the
testing
of
the
primary
endpoint,
in-stent
late
loss
at
9
months,
and the
secondary
endpoint,
12-month
TLF.
For
intent-to-treat
analyses,
all
TAXUS
PERSEUS
Small
Vessel
subjects
who
signed
the
IRB/EC-approved
written
Informed
Consent
Form
and
are
enrolled
in
the
study are
included
in
the
analysis,
regardless
of
whether
or
not
the
TAXUS
Element
Stent
was
implanted.
b.
Subjects
who
died
prior
to
completion
of
follow-up
window
and
prior
to
completing
a
12-month
clinical
follow-up
visit
are
considered
censored
and
are
excluded
from
calculation
of
proportion
of
subjects
who
completed
clinical
follow-up
visit.
c.
Based
on
subjects
eligible
for
12-month
clinical
follow-up
(excludes deaths
within
395
days).
d.
Includes subjects
who
have
died
in
both
the
numerator
and
the
denominator;
based
on
intent-to-treat
analysis
set.
e.
Based
on
intent-to-treat analysis
set.
Abbreviations:
BMS:
bare
metal
stent
(Express);
ITT:
intent-to-treat;
TLF:
target
lesion
failure.
B2.
Study
Population
Demographics
and
Baseline
Parameters
Demographics:
The
IONTM
Stent
group had
a
higher
rate
of
prior congestive
heart
failure
(8.1%
versus 2.4%),
previous smoking (48.6%
versus
36.8%),
and
higher
baseline
ejection
fraction
(57.9±9.4
versus 55.0±9.2)
as
compared with
the
historical BMS
control group.
The
ION
Stent
group had
lower
rates
of
baseline
unstable
angina
(20.1%
versus
29.6%)
and
current
smoking
(13.6%
versus
22.2%).
Baseline
lesion
characteristics:
Differences
in
the
ION
Stent
group
compared
to the
historical BMS
control
group
included shorter
lesion
length
(11.7±5.1
versus
12.9±5.1),
lower
incidence
of
ACC/AHA
Type B2/C
lesions
(58.0%
versus
77.6%),
and
lower
incidence
of
tortuosity
(8.9%
versus
18.4%).
However,
baseline
RVD
and
MLD
were significantly lower
in
ION
Stent
subjects
than
in
the
BMS
historical
control
(2.08±0.28 versus 2.19±0.35
for
RVD
and
0.55±0.23 versus
0.62±0.24
for
MLD,
respectively).
These
differences
were
not
expected
to
affect
outcomes variables,
as
propensity
score
adjustments showed
no change
in
the
outcome or conclusion
of
the
trial.
Baseline
Demographic
and Clinical
Characteristics,
ITT,
All
Subjects,
PERSEUS
SV
(N=349)
Historical Control
ION
BMS
(N=125)
(N=224)
Male
60.8% (76/125) 63.8%
(143/224)
64.2±10.9
(125)
64.7±10.3
(224)
Age(yr)
(41.0,
85.0)
(37.0,
85.0)
Ethnicity
and
Race
Hispanic
or
Latino
2.4%
(3/125)
4.9%
(11/224)
Caucasian
92.0%
(115/125)
91.1%
(204/224)
Asian
0.0%
(0/125)
0.0%
(0/224)
Black,
of
African heritage
4.0%
(5/125)
2.7%
(6/224)
Native
Hawaiian
or
other
NA
0.0%
(0/224)
Pacific Islander
American
Indian
or
0.0%
(0/125)
0.4%
(1/224)
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness Data
Page
40
A
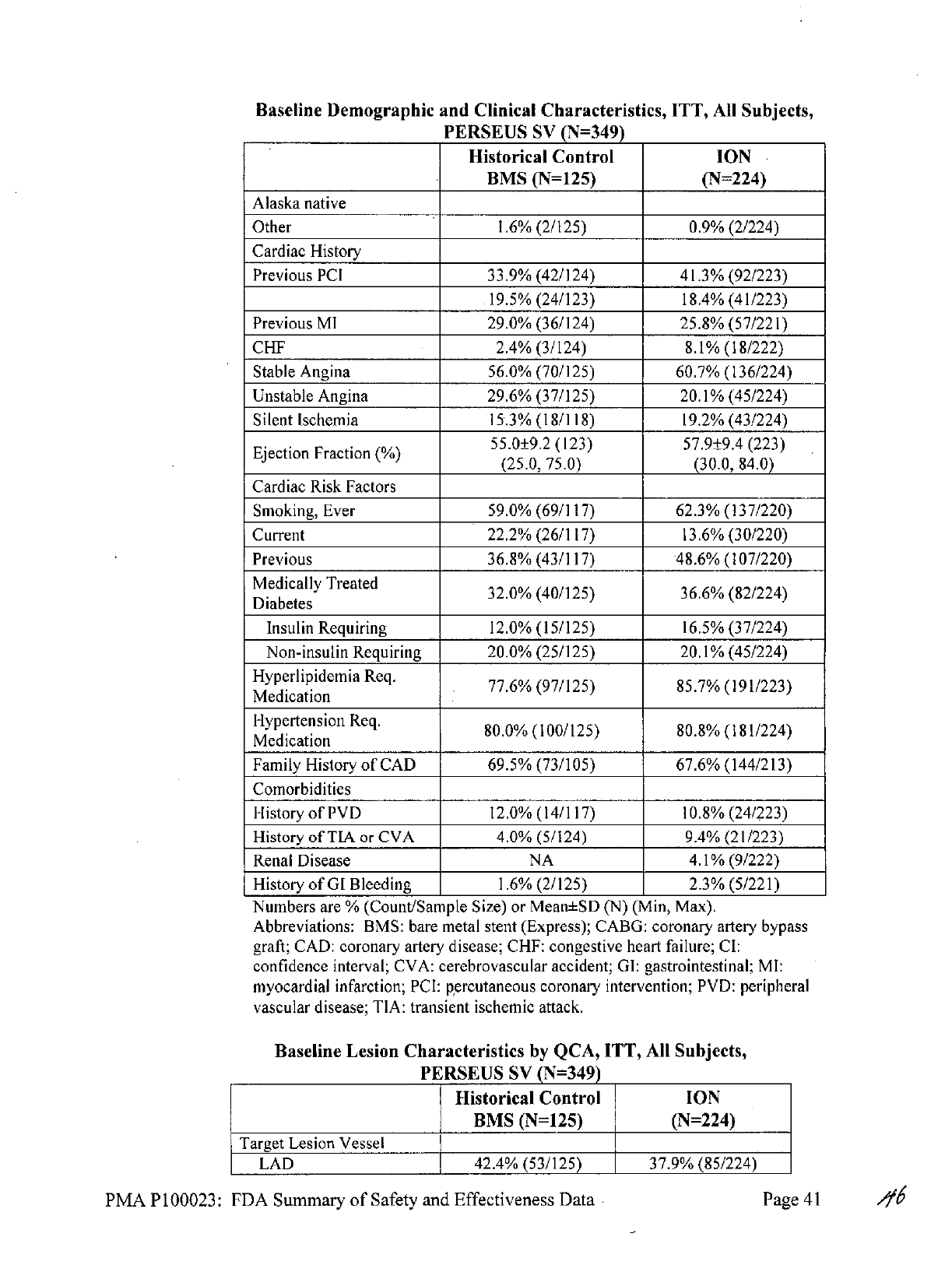
Baseline
Demographic
and
Clinical
Characteristics,
ITT,
All
Subjects,
PERSEUS
SV
(N=349)
Historical Control
ION
BMS
(N=125)
(N=224)
Alaska
native
Other
1.6%
(2/125)
0.9%
(2/224)
Cardiac History
Previous
PCI
33.9%
(42/124)
41.3%
(92/223)
19.5%
(24/123)
18.4%
(41/223)
Previous
MI
29.0%
(36/124)
25.8%
(57/221)
CHF
2.4%
(3/124)
8.1%
(18/222)
Stable
Angina
56.0%
(70/125)
60.7% (136/224)
Unstable Angina
29.6%
(37/125)
20.1%
(45/224)
Silent Ischemia
15.3%
(18/118)
19.2%
(43/224)
Fraction
(%)
55.0±9.2
(123)
57.9±9.4
(223)
(25.0,
75.0)
(30.0,
84.0)
Cardiac
Risk
Factors
Smoking,
Ever
59.0%
(69/117)
62.3%
(137/220)
Current
22.2% (26/117)
13.6%
(30/220)
Previous
36.8%
(43/117) 48.6%
(107/220)
Medically
Treated
32.0%
(40/125)
36.6%
(82/224)
Diabetes
Insulin
Requiring
12.0%
(15/125)
16.5%
(37/224)
Non-insulin
Requiring
20.0%
(25/125)
20.1%
(45/224)
Hyperlipidemnia
Req.
77.6%
(97/125)
85.7%
(191/223)
Medication
Hypertension
Req.
80.0%
(100/125)
80.8%
(181/224)
Medication
Family History
of
CAD
69.5%
(73/105) 67.6%
(144/213)
Comorbidities
History
of
PVD
12.0%
(14/117)
10.8%
(24/223)
History
of
TIA
or
CVA
4.0%
(5/124)
9.4% (21/223)
Renal
Disease
NA
4.1%
(9/222)
History
of
GI
Bleeding
1.6%
(2/125)
2.3%
(5/221)
Numbers
are
%
(Count/Sample
Size)
or
Mean+SD
(N)
(Min,
Max).
Abbreviations:
BMS:
bare
metal
stent
(Express);
CABG:
coronary
artery
bypass
graft;
CAD:
coronary
artery
disease;
CHF:
congestive
heart
failure;
Cl:
confidence
interval;
CVA:
cerebrovascular
accident;
GI:
gastrointestinal;
MI:
myocardial
infarction;
PCI:
percutaneous
coronary
intervention;
PVD:
peripheral
vascular
disease;
TIA:
transient
ischemic
attack.
Baseline Lesion
Characteristics
by
QCA,
ITT,
All
Subjects,
PERSEUS
SV
(N=349)
Historical
Control
ION
BMS
(N=125)
(N=224)
Target
Lesion Vessel
LAD
42.4%
(53/125)
37.9%
(85/224)
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
41

Baseline
Lesion
Characteristics
by
QCA,
ITT,
All
Subjects,
PERSEUS
SV
(N=349)
Historical
Control
ION
BMS
(N=125)
(N=224)
LCX
36.0%
(45/125)
41.5%
(93/224)
RCA
21.6% (27/125)
20.5%
(46/224)
Lesion
Location
Ostial
4.0%
(5/125)
6.3%
(14/224)
Proximal
33.6%
(42/125)
40.6%
(91/224)
Mid
44.8%
(56/125)
44.6%
(100/224)
Distal
17.6%
(22/125)
8.5%
(19/224)
Reference
Vessel
Diameter
2.2±0.4
(125)
2.1±0.3
(224)
(RVD,
mm)
(1.2,3.1)
(1.4,3.0)
Minimum
Lumen
Diameter
0.6±0.2
(125)
0.6±0.2
(224)
(MLD,
mm)
(0.2,
1.4)
(0.0,
1.2)
Percent
Diameter
Stenosis
71.5±10.4
(125)
73.5±10.2
(224)
(%
DS)
(40.5,
90.0)
(49.4,
100.0)
Lesion
Length
(mm)
12.9±5.1
(125)
11.7±5.1
(224)
(5.0,
25.7)
(3.8,
29.8)
Eccentric
Lesion
62.4%
(78/125)
66.5%
(149/224)
Bend
(degrees)
> 45
12.0%
(15/125)
6.3%
(14/224)
>90
0.0%
(0/125)
0.0%
(0/224)
Tortuosity,
Any
18.4%
(23/125)
8.9%
(20/224)
Moderate
13.6%
(17/125)
8.0%
(18/224)
Severe
4.8%
(6/125)
0.9%
(2/224)
Calcification,
Any
20.8% (26/125)
13.4%
(30/224)
Moderate
17.6%
(22/125) 9.4%
(21/224)
Severe
3.2%
(4/125)
4.0% (9/224)
Total Occlusion
0.8%
(1/125)
1.3%
(3/224)
Bifurcation
21.6%
(27/125)
23.7%
(53/224)
Sidebranch Stenosis
16.0±21.2
(27)
18.1±24.6
(53)
(0.0,
80.0)
(0.0,
90.0)
Modified
ACC/AHA
A
5.6%
(7/125)
9.4%
(21/224)
B
16.8%
(21/125)
32.6%
(73/224)
B2
58.4%
(73/125)
44.2%
(99/224)
C
19.2%
(24/125)
13.8%
(31/224)
Pre-Procedure
TIMI
Flow
0
0.0%
(0/125)
0.4%
(1/224)
1
0.8%
(1/125)
0.9%(2/224)
2
5.6%
(7/125)
0.4%
(1/224)
3
93.6%
(117/125)
98.2%
(220/224)
Numbers
are
%
(Count/Sample
Size)
or
Mean±SD
(N)
(Min, Max).
Abbreviations:
ACC/AHA:
American
College
of
Cardiology/American
Heart
Association;
BMS: bare
metal
stent
(Express);.CI:
confidence
interval;
DS:
diameter
stenosis;
LAD:
left
anterior
descending;
LCX: left
circumflex;
MLD:
minimum
lumen
diameter;
QCA:
quantitative
coronary
angiography;
RCA:
right coronary
artery;
RVD:
reference
vessel
diameter;
TIMI:
thrombolysis
in
myocardial infarction.
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
42

B3.
Safety
and
Effectiveness
Results
1.
Safety
Results
The
analysis
of
safety was
based
on
the
ION
stent cohort
of
224
subjects
available
for
the
9-month primary
endpoint.
The
principal adverse
events
for
this
study
are
presented
below
in
Table
20.
Table
20:
Principal
Adverse
Events
PERSEUS
Small
Vessel
to
1
Year
Express
ION
Stent
Stentb
(N=224)
(N=125)
In-Hospital
MACE
0.0%
(0/224)
1.6%
(2/125)
30-Day
MACE 0.9%
(2/221)
2.4%
(3/124)
9-Month
MACE
7.8%
(17/218)
14.6%
(18/123)
Cardiac
Death
0.9%
(2/218)
0.8%
(1/123)
MI
0.9%
(2/218)
2.4%
(3/123)
Q-Wave
MI
0.5%
(1/218)
0.0%
(0/123)
Non-Q-Wave
MI
0.5%
(1/218)
2.4%
(3/123)
TVR
6.9%
(15/218)
12.2%
(15/123)
TLR
3.7%
(8/218)
10.6%
(13/123)
Non-TLR
5.0%
(11/218)
4.1%
(5/123)
1-Year
MACE
12.4%
(27/218)
27.3%
(33/121)
Cardiac
Death
1.4%
(3/218)
0.8%
(1/121)
MI
0.9%(2/218)
2.5%
(3/121)
Q-Wave
MI
0.5%
(1/218)
0.0%
(0/121)
Non-Q-Wave
MI
0.5%
(1/218)
2.5%
(3/121)
TVR
11.5%
(25/218)
24.8%
(30/121)
TLR
6.0%
(13/218)
20.7%
(25/121)
Non-TLR
7.8%
(17/218)
7.4%
(9/121)
1-Year
ARC
Stent
Thrombosis
Definite
or
Probable
0.5%
(1/215)
0.8% (1/119)
Definite
0.5%
(1/215)
0.8%(1/119)
Probable
0.0%(0/215)
0.0%
(0/119)
b
DES
Control
2.
Effectiveness
Results
The
analysis
of
effectiveness
was
based on
the
224
evaluable
subjects
at
the
9-
month primary
endpoint.
Key
safety
and
effectiveness
outcomes
are
presented
in
Tables
21
through
26.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
43
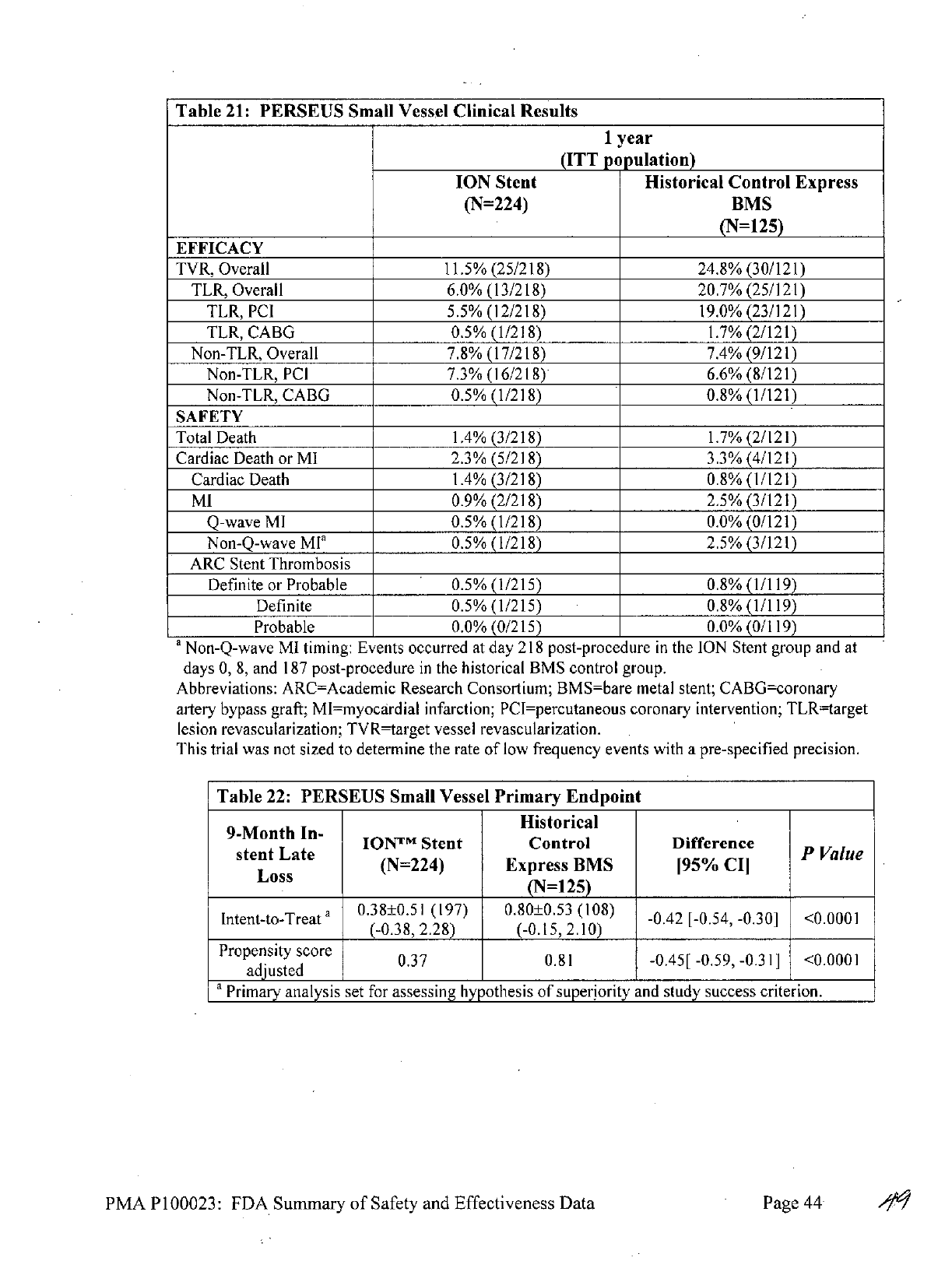
Table
21:
PERSEUS
Small
Vessel
Clinical
Results
1
year
(ITT
population)
ION
Stent
Historical Control
Express
(N=224)
BMS
(N=125)
EFFICACY
TVR, Overall
11.5%
(25/218)
24.8%
(30/121)
TLR,
Overall
6.0%
(13/218)
20.7%
(25/121)
TLR,
PCI
5.5%
(12/218)
19.0%
(23/121)
TLR,
CABG
0.5%
(1/218)
1.7%
(2/121)
Non-TLR,
Overall
7.8%
(17/218)
7.4%
(9/121)
Non-TLR,
PCI
7.3%
(16/218)
6.6%
(8/121)
Non-TLR,
CABG
0.5%
(1/218)
0.8%
(1/121)
SAFETY
Total
Death
1.4%
(3/218)
1.7%
(2/121)
Cardiac
Death
or
MI
2.3%
(5/218)
3.3%
(4/121)
Cardiac
Death
1.4%
(3/218)
0.8%
(1/121)
MI
0.9%
(2/218)
2.5%
(3/121)
Q-wave
MI
0.5%
(1/218)
0.0%
(0/121)
Non-Q-wave
MI'
0.5%
(1/218)
2.5%
(3/121)
ARC
Stent Thrombosis
Definite
or
Probable
0.5%
(1/215)
0.8% (1/119)
Definite
0.5%
(1/215)
0.8%
(1/119)
Probable
0.0%
(0/215)
0.0%
(0/119)
a
Non-Q-wave
MI
timing: Events occurred
at
day
218
post-procedure
in
the
ION
Stent
group
and
at
days
0,
8,
and
187
post-procedure
in
the
historical
BMS
control
group.
Abbreviations:
ARC=Academic
Research
Consortium; BMS=bare
metal
stent;
CABG=coronary
artery
bypass
graft;
MI=myocardial
infarction;
PCI=percutaneous
coronary intervention;
TLR-target
lesion
revascularization;
TVR=target
vessel
revascularization.
This
trial
was not sized to
determine
the
rate
of
low
frequency events
with
a
pre-specified
precision.
Table
22:
PERSEUS
Small
Vessel
Primary
Endpoint
Historical
9-Mnth
Ie
IOM
Tm
Stent
Control
Difference
P
Value
sse
L(N=224)
Express
BMS
195%
CII
(N=125)
Intent-to-Treat
a
038±0.51
(197)
0.80±0.53
(108) -0.42
[-0.54,
-0.30]
<0.0001
(-0.38,
2.28)
(-0.15,
2.10)
Propensity
score
0.37
0.81
-0.45[
-0.59,
-0.31]
<0.000
1
adjusted
I
I
I
a
Primary
analysis
set
for
assessing
hypothesis
of
superiority
and
study
success criterion.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
44
-

Table
23:
PERSEUS
Small
Vessel
Secondary
Endpoint
Performance
ION
Stent
Upper
95%
12-Month
TLF
Confidence
P
Value
Goal
(N=224)Limit
Intent-to-Treata
19.5%
7.34%
10.80%
<0.0001
1_
(16/218)
a
Primary
analysis
set
for
assessing
hypothesis
of
superiority
and
study
success
criterion.
Table
24:
PERSEUS
Small
Vessel
Angiographic
Results
Historical Control
N=197
teExpress
BMS
Angiographic
Outcomes
a
(N=108)
MLD
(mm),
In-stent
Post-Procedure
2.11±0.21
(197)
2.09±0.30
(108)
9-Month
1.73±0.53
(197)
1.29±0.55
(108)
MLD
(mm),
Analysis
Segment
Post-Procedure
1.70±0.29
(197)
1.76±0.38
(108)
9-Month
1.50±0.48
(197)
1.22±0.50
(108)
Acute
Gain
(mm),
In-stent
1.57±0.27
(197)
1.47±0.33
(108)
Acute
Gain,
Analysis
Segment
(mm)
1.16±0.30
(197)
1.14±0.39
(108)
%
DS,
In-stent
Post-Procedure
0.31±10.76
(197)
6.63±10.97
(108)
9-Month
18.48±23.31
(197)
40.72±23.64
(108)
%
DS,
Analysis
Segment
Post-Procedure
20.12±9.42
(197)
22.34±10.69
(108)
9-Month
29.82±19.82
(197)
43.85±21.44
(108)
Late
Loss,
In-stent
(mm)
0.38±0.51
(197)
0.80±0.53
(108)
Late
Loss,
Analysis
Segment
(mm)
0.21±0.41
(197)
0.53±0.52
(108)
Binary
Restenosis
In-stent
restenosis
11.7%
(23/197)
34.3%
(37/108)
Analysis
segment
restenosis
13.7%
(27/197)
38.0%
(41/108)
a
Includes
all
subjects
with
paired
lesion
data.
Abbreviations:
BMS=bare
metal
stent;
DS=diameter
stenosis;
MLD=minumum
lumen
diameter.
Table
25:
PERSEUS
Small
Vessel
Stent
Thrombosis
Historical
Control
N=2
StntExpress
BMS
Intent-to-Treat
Population
(N=125)
Protocol
Defined
Stent
Thrombosis
Cumulative
through
I
year
0.5%
(1/215)
0.8%
(1/119)
Acute
ST
(<24
hrs)
0.0%
(0/224)
0.0%
(0/125)
Subacute
ST
(>24
hrs
and
530
days)
0.0%
(0/221)
0.8%
(1/125)
Late
ST
(>30
days
and
12
months)
0.5%
(1/217)
0.0%
(0/124)
ARC
Definite
&
Probable
Stent
Thrombosis
Cumulative
through
1
year
0.5%
(1/215)
0.8% (1/119)
Acute
ST
(524
hrs)
0.0%
(0/224)
0.0%
(0/125)
Subacute
ST
(>24
hrs
and
530 days)
0.0%
(0/221)
0.8%
(1/125)
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
45
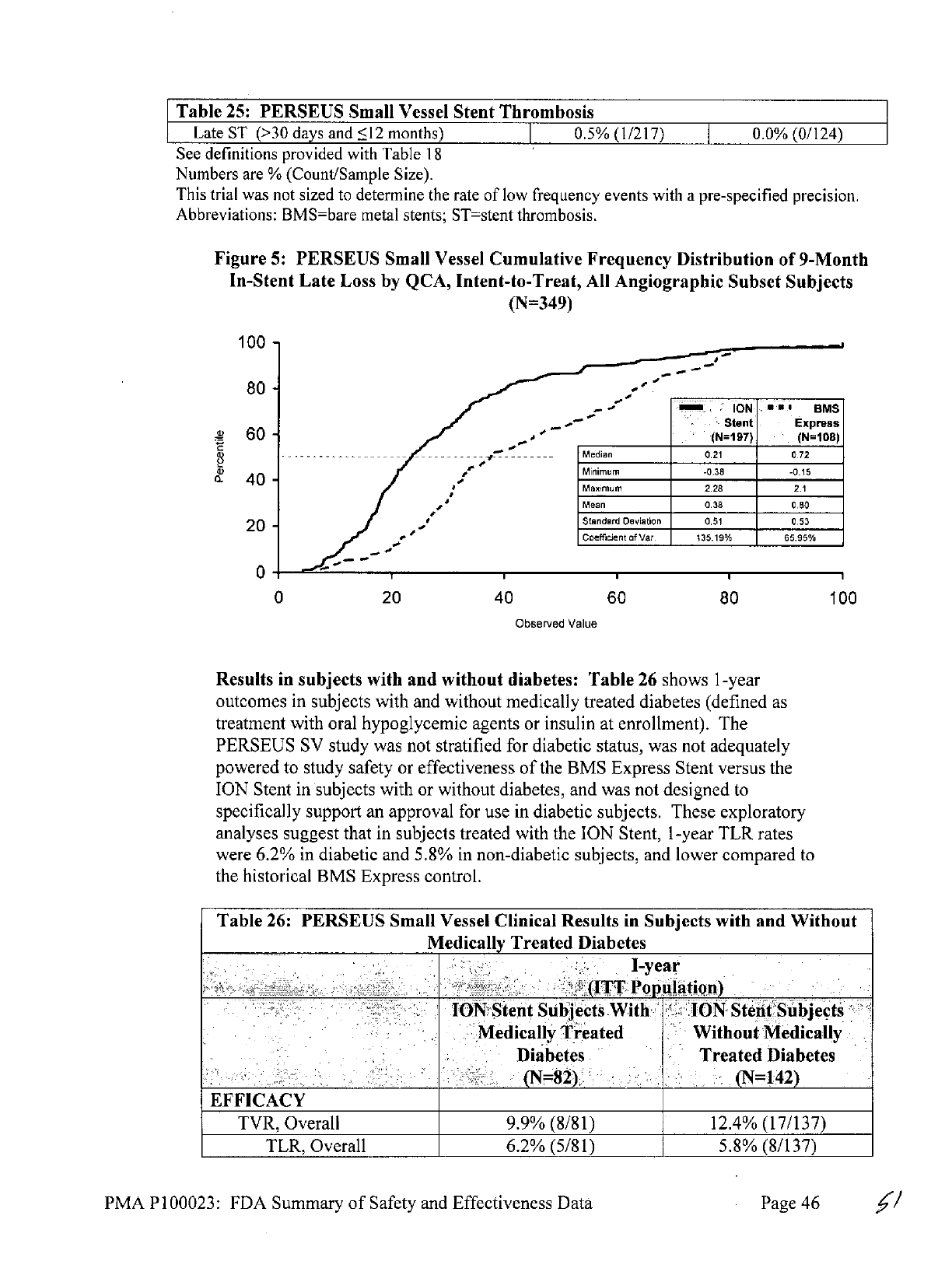
Table
25:
PERSEUS
Small
Vessel
Stent
Thrombosis
Late
ST
(>30
days
and
<12
months)
0.5%
(1/217)
0.0%
(0/124)
See
definitions
provided
with Table
18
Numbers
are
%
(Count/Sample
Size).
This
trial
was
not
sized
to
determine
the
rate
of
low
frequency
events
with
a
pre-specified
precision.
Abbreviations:
BMS=bare
metal
stents;
ST=stent
thrombosis.
Figure
5:
PERSEUS
Small
Vessel
Cumulative
Frequency
Distribution
of
9-Month
In-Stent
Late
Loss
by
QCA,
Intent-to-Treat,
All
Angiographic
Subset
Subjects
(N=349)
100
-
80 -______
_o
*** sus
--
Stent
Express
_
60
-
___
_
(N=197)
.
(N=108)
------------ .----
Median
0.21 0.72
1
Minimum
-0.38
-0.15
40
-
Maximum
2.28
2.1
Mean
0.38
0.80
20
Standard
Deviation
0.51
0.53
Coefficient
of
Var.
135.19%
65.95%
0
0
20
40
60
80
100
Observed
Value
Results
in
subjects
with
and
without
diabetes:
Table
26
shows
1-year
outcomes
in
subjects with
and
without medically
treated
diabetes (defined
as
treatment
with
oral
hypoglycemic
agents
or
insulin
at
enrollment).
The
PERSEUS
SV
study
was
not
stratified
for
diabetic status,
was
not adequately
powered
to
study safety
or
effectiveness
of
the
BMS
Express
Stent
versus the
ION
Stent
in
subjects
with
or
without
diabetes,
and
was
not
designed
to
specifically support
an
approval
for use
in
diabetic
subjects.
These
exploratory
analyses
suggest
that
in
subjects
treated with
the
ION
Stent,
1-year
TLR
rates
were
6.2%
in
diabetic
and
5.8%
in
non-diabetic
subjects,
and
lower
compared
to
the
historical
BMS
Express
control.
Table
26:
PERSEUS
Small
Vessel
Clinical
Results
in
Subjects
with
and
Without
Medically
Treated
Diabetes
I-year
(ITT
Population)
ION
Stent
Subjects
With
ION
Stent
Subjects
Medically
Treated
Without
Medically
Diabetes
Treated
Diabetes
(N=82)
(N=
142)
EFFICACY
TVR,
Overall
9.9%
(8/81)
12.4%
(17/137)
TLR,
Overall
6.2%
(5/81) 5.8%
(8/137)
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
46

Table
26:
PERSEUS
Small
Vessel
Clinical
Results
in
Subjects with
and
Without
Medically
Treated
Diabetes
I-year
(ITT
Population)
ION
Stent
Subjects
With
ION
Stent
Subjects
Medically
Treated
Without
Medically
Diabetes
Treated
Diabetes
(N=82)
(N=142)
TLR,
PCI
6.2%(5/81)
5.1%
(7/137)
TLR,
CABG
0.0%(0/81)
0.7%
(1/137)
Non-TLR,
Overall
6.2%
(5/81)
8.8%
(12/137)
Non-TLR,
PCI
6.2%
(5/81)
8.0%
(11/137)
Non-TLR,
CABG
0.0%
(0/81)
0.7%
(1/137)
SAFETY
Total
Death
2.5%(2/81)
0.7%
(1/137)
Cardiac
Death
or
MI
3.7%
(3/81)
1.5%
(2/137)
Cardiac
Death
2.5%
(2/81)
0.7%
(1/137)
MI
1.2%(1/81)
0.7%
(1/137)
Q-wave
MI
1.2%(1/81)
0.0%
(0/137)
Non-Q-wave
MI
0.0%
(0/81)
0.7%
(1/137)
ARC
Stent
Thrombosis
Definite
or
Probable
1.3%
(1/79)
0.0%
(0/136)
Definite
1.3%(1/79)
0.0%
(0/136)
Probable
0.0%(0/79)
0.0%
(0/136)
This
trial
was
not
sized
to
determine
the rate
of
low
frequency
events
with
a
pre-specified
precision.
Abbreviations:
ARC=Academic
Research
Consortium;
CABG=coronary
artery
bypass
graft;
DES=drug-eluting
stent;
MI=myocardial
infarction;
PCI=percutaneous
coronary intervention;
TLR-target
lesion
revascularization;
TVR=target
vessel
revascularization.
C.
PERSEUS
Clinical
Trial
Program
-
Sex/Gender
Analyses
PERSEUS
Workhorse
To
evaluate
for
possible
sex-based
differences
in
outcome
of
treatment
with
the
ION
Stent,
sex/gender-specific
analyses
were
performed
on
safety
and
effectiveness
endpoints.
The
results
suggest
that
the
general
conclusions
of
the
overall
study
regarding
both
safety
and
effectiveness
can
be
generalized
for
males
and
females.
In
the
PERSEUS
Workhorse
ITT
population,
of
the
942
subjects
randomized
to
ION,
667
subjects
were male
(70.8%)
and
275
subjects
were
female
(29.2%).
The
proportions
in
the
TAXUS
Express
group
were
similar
(68.8%
male,
31.2%
female).
In
comparison,
the
prevalence
of
coronary
artery
disease
(CAD)
is
estimated
at
9.2
million
in
males
and
8.4
million
in
females
for
adults
age
20
and
older
the
United
States
(i.e.,
the
CAD
population
is
estimated
to be
52.2%
males
and
47.7%
females).
The
disproportionate
enrollment
distribution
in
this
trial
may
be
partly
attributable
to
gender
differences
in
symptoms
and
pathophysiology
5
,
which
may
lead
to
under-
diagnosis
and
referral
of
female
subjects
with
CAD.
The
gender
proportions
enrolled
in
this
trial
are
similar
to
other
drug-eluting
stent
trials;
a
meta-analysis
of
paclitaxel-
eluting
stent clinical
trials
reported
an
overall
gender
distribution
of
71.8%
male
and
28.2%
female.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
47

The
PERSEUS
WH
study
was
not powered
to
study
safety
or effectiveness
of
the
ION
Stent
versus
the
TAXUS
Express
Stent
in
sex-specific
subgroups.
PERSEUS
WH
primary
and
secondary
endpoint
data
were
assessed
for
differences
between
male
and
female
subgroups,
as
well
as
for
any
interaction
between
treatment
group
and
gender.
These
post
hoc
analyses
suggest
that
in
subjects
treated
with
the
ION
stent,
12-month
TLF rates
were
7.0%
in
females
and
5.1%
in
males,
and
9-month
%DS
was
26.95±18.62
in
females
and
25.71±17.32
in
males.
Numerical
differences
were
observed
in
the
treatment
effect
(i.e.,
the
difference
between
the
ION
Stent
and
TAXUS
Express
Stent),
as
shown
in
Table
27
below.
No
significant
treatment-by-gender
interaction
effect
was
observed
for the
primary
endpoint
of
12-month TLF
(P=0.5485).
A
marginally
significant
treatment-by-gender
interaction
effect
was
observed
for
the
secondary
endpoint
of
9-month
in-segment
%DS
under
the
natural
log
transformation
(P=0.0628).
However,
this analysis
is
limited
by
the small
sample
size;
fifteen
(15)
female
TAXUS
Express
subjects
have
available
9-month
in-segment
%DS
data,
and
the
mean
%DS
for
those subjects
was
markedly
low.
Considering
the small
sample
size
and
the
lack
of
observed
interaction
effect
for
the
primary
endpoint
of
12-month
TLF,
there
does
not
appear
to
be
a
clinically
significant
treatment-by-gender
interaction
in
the
PERSEUS
WH
trial.
This
suggests
that
the
overall
conclusions
of
this
trial
regarding
both
safety
and
effectiveness
of
the
ION
Stent
can
be
generalized
for
males
and
females.
Table
27:
PERSEUS
Workhorse
Primary
and
Secondary
Endpoint
Results
by
Gender,
Intent-to-Treat,
All
Subjects
(N=887)
Relative
Risk
Difference
Interactio
TAXUS
Express
ION
Stent
[95%
Cl]
[95%
Cl]
P
value
n
p-Value
12-month
TLF
(Primary
Endpoint)
(N=220)
(N=667)
_______
-
(N=22)
(N667)
1.09
[0.54,
0.4%
[-2.9%,
Male
4.7%
(10/214)
5.1%
(33/650)
.17]
3%]
0.8136
(N=100)
(N=275)
21]37]058
(N=10)
(N=75)
-0.77
[0.36,
-2.1%
[-8.5%,
Female
9.1%(9/99)
7.0%
(19/272)
1.6
4%]
0.4971
1.64]
4.3%]
9-month
Percent
Diameter
Stenosis
In-Segment
(Secondary
Endpoint)
(N=53)
(N=180)
28.57±19.16
(46)
25.71±17.32
Male
(7.56,
100.00)
(156)
NA
-2.86
[-8.70,
2.97]
0.3373
(3.31,
100.00)
0.0628
(N=21)
(N=76)
26.95±18.62
Female
19.71
1)
(72)
NA
7.34
[-2.29,
16.97]
0.1389
(9.71,
31.66)
(42,9.9
______
_
______
(4.22,
95.29)
Table
28
shows
12-month
clinical
results
in
male
and
female
subjects.
PMA P100023:
FDA
Summary
of
Safety and
Effectiveness Data
Page
48
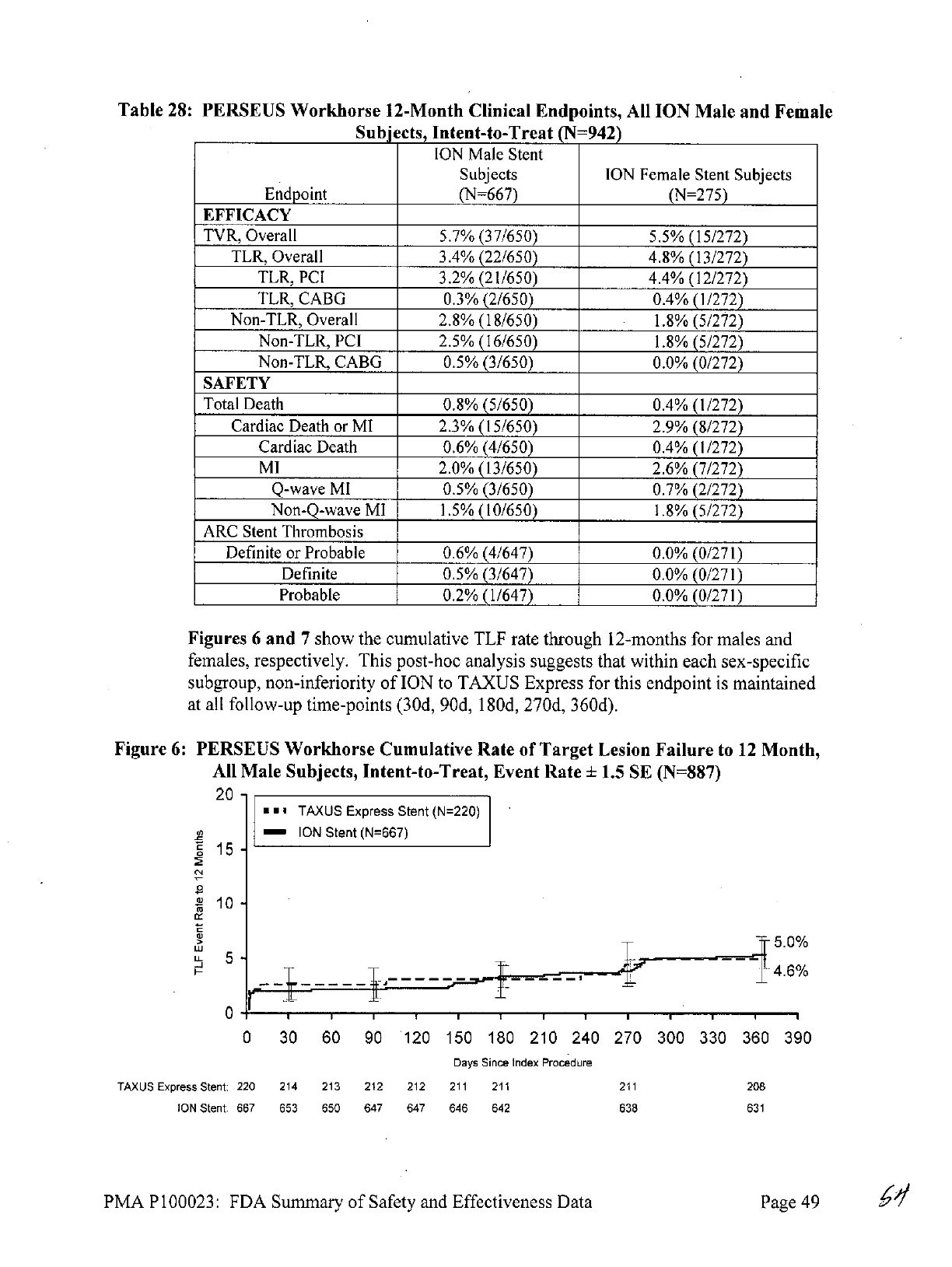
Table
28:
PERSEUS
Workhorse
12-Month
Clinical
Endpoints,
All
ION Male
and
Female
Subjects,
Intent-to-Treat
(N=942)
ION
Male
Stent
Subjects
ION
Female
Stent Subjects
Endpoint
(N=667)
(N=275)
EFFICACY
TVR,
Overall
5.7%
(37/650)
5.5%
(15/272)
TLR,
Overall
3.4%
(22/650)
4.8%
(13/272)
TLR,
PCI
3.2%
(21/650)
4.4%
(12/272)
TLR,
CABG
0.3%
(2/650)
0.4%
(1/272)
Non-TLR,
Overall
2.8%
(18/650)
1.8%
(5/272)
Non-TLR,
PCI
2.5%
(16/650)
1.8%
(5/272)
Non-TLR,
CABG
0.5%
(3/650)
0.0%
(0/272)
SAFETY
Total
Death
0.8%
(5/650)
0.4%
(1/272)
Cardiac
Death
or MI
2.3%
(15/650)
2.9%
(8/272)
Cardiac
Death
0.6%
(4/650)
0.4%
(1/272)
MI
2.0% (13/650)
2.6%
(7/272)
Q-wave
MI
0.5%
(3/650)
0.7%
(2/272)
Non-Q-wave
MI
1.5%
(10/650)
1.8%
(5/272)
ARC Stent
Thrombosis
Definite
or
Probable
0.6%
(4/647)
0.0%
(0/271)
Definite
0.5%
(3/647)
0.0%
(0/271)
Probable
0.2%
(1/647)
0.0%
(0/271)
Figures
6
and
7
show
the
cumulative
TLF
rate
through
12-months
for males
and
females,
respectively.
This
post-hoc
analysis
suggests
that within
each
sex-specific
subgroup,
non-inferiority
of
ION
to
TAXUS
Express
for
this
endpoint
is
maintained
at
all
follow-up
time-points
(30d,
90d,
180d,
270d,
360d).
Figure
6:
PERSEUS
Workhorse
Cumulative
Rate of
Target
Lesion
Failure
to
12
Month,
All
Male
Subjects,
Intent-to-Treat,
Event
Rate
+
1.5
SE
(N=887)
20
-
TAXUS
Express
Stent
(N=220)
ION
Stent
(N=667)
E
15
-
10
>
5.0%
w
u_
5
4.6%
0
30
60
90
120 150
180
210
240
270
300
330
360
390
Days
Since
Index Procedure
TAXUS
Express
Stent:
220
214 213 212 212
211 211 211
208
ION
Stent: 667
653
650
647 647 646
642
638
631
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
49

Figure
7:
PERSEUS
Workhorse
Cumulative
Rate of
Target
Lesion
Failure
to
12
Month,
All
Female
Subjects,
Intent-to-Treat,
Event
Rate
+
1.5
SE
(N=375)
20
-
TAXUS
Express
Stent
(N=100)
ION
Stent
(N=275)
g 15
C
>
6.9%
LLL
10
-
-
-
.2
0
30 60
90
120 150
180
210
240
270
300
330
360
390
Days Since
Index
Procedure
TAXUS
Express
Stent:
100
97
97 97
96
96
95
94
93
ION
Stent: 275
268
268 267
266
264
263
262
256
PERSEUS
Small
Vessel
To
evaluate
for
possible
sex-based differences
in
outcome
of
treatment
with
the
ION
Stent
when used
in
small
vessels,
sex/gender-specific
analyses
were
performed
on
safety
and
effectiveness
endpoints.
The
results
suggest
that
the
general
conclusions
of
the
overall
study
regarding
both
safety
and
effectiveness
can
be
generalized
for
males
and
females.
In
the
PERSEUS
Small
Vessel
ITT
population,
of
the 224
registry
subjects,
143
subjects
were
male
(63.8%)
and
81
subjects
were
female
(36.2%).
The
proportions
in
the
BMS
Express
historical
control
group
were
similar
(60.8%
male,
39.2%
female).
In
comparison,
the
prevalence
of
coronary
artery
disease
(CAD)
is
estimated
at
9.2
million
in
males
and
8.4
million
in
females
for
adults
age
20
and
older
the
United
States
(i.e.,
the
CAD
population
is
estimated
to be
52.2%
males
and
47.7%
females).
The
disproportionate
enrollment
distribution
in
this trial may
be
partly
attributable
to
gender
differences
in
symptoms
and
pathophysiology
5
,
which
may
lead
to
under-
diagnosis
and
referral
of
female
subjects
with
CAD.
The
gender
proportions
enrolled
in
this
trial
are
somewhat
more
representative
of
the
disease prevalence
than
the
Workhorse
trial
(70.8%
male;
29.2%
female)
and
other
drug-eluting
stent
trials.
This
may
be
due in
part
to
the
smaller
average
reference
vessel
diameter
in
female
subjects
6
.
The
PERSEUS
SV
study
was
not
powered
to
study
safety
or effectiveness
of
the
ION
Stent
in
sex-specific
subgroups.
PERSEUS
SV
primary
and
secondary
endpoint
data
were
assessed
for
differences
between
male
and
female
subgroups,
as
well
as
for
any
interaction
between
treatment
group
and
gender.
These
post
hoc
analyses
suggest
that
in
subjects
treated
with
the
ION
stent
in
small
vessels,
9-month
in-segment
late
loss was
0.41+0.48
in
females
and
0.36±0.52
in
males,
and
12-month TLF
was
5.0%
in
females
and
8.7%
in
males.
In
the
BMS
Express
historical
control
group,
rates
of
12-month
TLF
were also
numerically
higher
in
males
(25.2%)
than
females
(16.8%).
These
observations
are
limited
by
the
small
sample
size
available
for
these analyses.
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
50

Treatment
effect
(i.e.,
superiority
of
ION
Stent
to
historical
control
BMS)
was
demonstrated
for
both
males
and
females,
as
shown
in
Table
29
below.
No
significant
treatment-by-gender
interaction
effect
was
observed
for the
primary
endpoint
of
9-month
in-stent
late
loss
(P=0.7255)
or
12-month
TLF
(P=0.9246).
This
suggests
that
the
overall
conclusions
of
this
trial
regarding
both
safety
and
effectiveness
of
the
ION
Stent
in
small
vessels
can
be
generalized
for
males
and
females.
Table
29:
PERSEUS
Small
Vessel
Primary
and
Secondary
Endpoint
Results,
by
Gender,
Intent-to-Treat,
All
Subjects
(N=219)
Historical
Relative
Risk
Difference
Interactio
Control
BMS
ION
Stent
[95%
CI]
[95%
CI]
P
value
n
p-Value
9-month
Late
Loss
In-Stent
(Primary
Endpoint)
(N=76)
(N=143)
0.36±0.52
Male
08±54
(0)
(127)
NA
-0.44
[-0.59,
-0.28]
<.0001
(-0.38, 2.28)
0.7255
(N=49)
(N=81)
Female
0.80±0.50
(38)
0.41±0.48
(70)
NA
-0.39
[-0.59,
-0.20]
0.0001
(-0.09,
2.10)
(-0.34,
1.59)
1
12-month
TLF
(Secondary
Endpoint)
(N=76)
(N=143)
Male
25.7%
(19/74)
8.7%
(12/138)
0.34
[0.17,
0.66]
-17.0%
[-28.0%,
-6.0%]
0.0009
0.9246
(N=49)
(N=81)
Female
17.0%
(8/47)
5.0%
(4/80)
0.29
[0.09,
0.92]
-12.0%
[NA]
0.0549*
Table
30
shows
12-month
clinical
results
in
male
and
female
subjects.
Table
30:
PERSEUS
Small
Vessel
12-Month
Clinical
Endpoints,
All
ION
Male
and
Female
Subjects,
Intent-to-Treat
(N=224)
ION
Male
Stent
Subjects
ION
Female
Stent
Subjects
Endpoint
(N=143)
(N=81)
EFFICACY
TVR,
Overall
13.8%
(19/138)
7.5%
(6/80)
TLR,
Overall
7.2%
(10/138)
3.8%
(3/80)
TLR,
PCI
6.5%
(9/138)
3.8%
(3/80)
TLR,
CABG
0.7%
(1/138)
0.0%(0/80)
Non-TLR,
9.4%
(13/138)
5.0%
(4/80)
Overall
PCI
Non-TLR,
8.7%
(12/138)
5.0%(4/80)
CABG
Non-TLR,
0.7%
(1/138)
0.0%(0/80)
SAFETY
Total Death
1.4%
(2/138)
1.3%(1/80)
Cardiac
Death
or
MI
2.9%
(4/138)
1.3%
(1/80)
Cardiac
Death
1.4%
(2/138)
1.3%
(1/80)
MI
1.4%
(2/138)
0.0%(0/80)
Q-wave
MI
0.7%
(1/138)
0.0%
(0/80)
PMA P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
51

ION
Male
Stent
Subjects
ION
Female
Stent
Subjects
Endpoint
(N=143)
(N=81)
MI
Non-Q-wave
0.7%
(1/138)
0.0%
(0/80)
ARC Stent
Thrombosis
Definite
or
Probable
0.7%
(1/136)
0.0%
(0/79)
Definite
0.7%
(1/136)
0.0%(0/79)
Probable
0.0%
(0/136)
0.0%
(0/79)
In
the
PERSEUS
SV
Trial, the study
success
criterion
for
12-month TLF
was
met
for
both
sexes
(greater
than
performance
goal
of
19.5%).
Figures
8
and
9
show
the
cumulative
TLF
rate
through
12-months
for
males
and
females,
respectively.
This
post-hoc
analysis
suggests
that within
each
sex-specific
subgroup,
the
ION
group
had
lower
TLF
rates
than
the BMS
Express
historical
control
group
at
all
follow-up
time-
points
(30d,
90d,
180d,
270d,
360d),
although
confidence
intervals
are
wide
and
overlap
at
earlier
time
points.
Figure
8:
PERSEUS
Small
Vessel
Cumulative
Rate
of
Target
Lesion
Failure
to
12
Month,
All
Male
Subjects,
Intent-to-Treat,
Event Rate
±
1.5
SE
(N=219)
30
BMS
Express
(N=76)
ION
Stent
(N=143)
:
25.2%
0
L
10
8.7%
U-
-J~
0
30
60
90
120
150 180
210 240 270
300
330
360
390
Days
Since
Index
Procedure
BMS:
76
76 76
76
74
72
71
71
67
ION
Stent:
143
141
138 138
138 137
137
136
130
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
52

Figure
9
PERSEUS
Small
Vessel
Cumulative
Rate
of
Target
Lesion
Failure
to
12
Month,
All
Female
Subjects,
Intent-to-Treat,
Event
Rate
±
1.5
SE
(N=130)
30
BMS
Express
(N=49)
ION
Stent
(N=81)
0
20
--
C
20
16.8%
0)
I
Ili
lI
S10
U-
-
7
5.0%
0
30 60
90
120 150
180
210
240 270
300
330 360
390
Days
Since
Index
Procedure
BMS:
49
46 46 45
45 44
43
43
41
ION
Stent:
81
79
79 79
79
79
79
79
78
X.
PANEL
MEETING
RECOMMENDATION
AND
FDA'S
POST-PANEL
ACTION
In
accordance
with
the
provisions
of
section
515(c)(2)
of
the
Act
as
amended
by
the
Safe
Medical
Devices
Act
of
1990,
this
PMA
was
not referred
to
the
Circulatory
Systems
Devices
Panel,
an
FDA
advisory
committee,
for
review
and
recommendation
because the
information
in
the
PMA
substantially
duplicates
information
previously
reviewed
by
this
panel.
XI.
CONCLUSIONS
DRAW
FROM
PRECLINICAL
AND
CLINICAL
STUDIES
The safety
and
effectiveness
of
the
ION
Paclitaxel-Eluting
Platinum
Chromium
Coronary
Stent
System
(Monorail
and
Over-The-Wire)
is
based
on the
results
obtained
from:
evaluation
of
biocompatibility;
characterization
of
in
vivo
pharmacokinetics;
in
vitro
engineering
testing;
coating
characterization;
chemistry,
manufacturing
and
controls
information;
in
vivo
animal
testing;
sterilization
information;
stability
testing;
and
clinical
studies.
These
tests revealed
the
following:
A.
Safety
Conclusions
The
biocompatibility,
in
vivo
pharmacokinetics,
and
in
vivo
animal
testing
conducted
demonstrate that
the
acute
and
chronic
in
vivo
performance
characteristics
of
the
product
provide
reasonable
assurance
of
safety
and
acceptability
for
clinical
use.
The
in
vitro
engineering
testing
conducted
on
the
stent
and
delivery
system(s)
demonstrated
that
the
performance characteristics
met
the
product
specifications
and
the
coating
characterization testing adequately
described
the
important attributes
of
the
paclitaxel/polymer
coating.
The
chemistry,
manufacturing,
and
controls information
ensures
that
product
meeting
specifications
will
be
released.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
53

The
test
results
obtained
from
the
sterilization
testing
demonstrated
that
the
product
can
be
adequately
sterilized
and
is
acceptable
for
clinical
use.
The
stability
testing
demonstrated
that
the
product
can
be
labeled
with
a
shelf
life
of
18
months.
The
results
of
the
PERSEUS
Clinical
Trial
Program
showed
that
the
principal
adverse
events
rates for
the
ION
were
similar
to
those
observed
for
the
TAXUS
Express
control
group.
Although
an
angiographic
core
laboratory
review
of
all
available
angiograms
revealed
a
total
of
3
stent
fractures
-
2
ION
stent
fractures
(Type
33
fractures
that
were
seen
on
angiograms
performed
at 286
and
259
days
post-stent
implantation,
respectively)
and
1
Taxus Express
stent fracture
(Type
43
fracture
noted
on
an
angiogram
performed
861
days
post-stent
implantation).
Only
the fracture
that
occurred
with the
Taxus Express
stent
was
associated
with
a
major
adverse
cardiovascular
event
(a
TLR).
B.
Effectiveness
Conclusions
PERSEUS
Workhorse
The
effectiveness
of
ION
workhorse
stents
was based
on the
rate
of
target
lesion
failure
(TLF)
at
12
months
post-index
procedure,
testing
non-inferiority
of
the
ION
Stent
relative
to
the
TAXUS
Express
Paclitaxel-Eluting
Stent
control.
In-segment
percent
diameter
stenosis
at
9
months
post-index
procedure
as
measured
by
quantitative
coronary
angiography
(QCA)
is
the
secondary
endpoint.
The
prespecified
Bayesian
analyses
showed
that
there
was
a
99.96%
Bayesian
posterior
probability
that
the
ION
stent
was
non-inferior
to
the
TAXUS
Express
stent,
demonstrating
non-inferiority
of
the
ION
stent
versus
the
TAXUS
Express
stent.
PERSEUS
Small
Vessel
The
effectiveness
of
the
ION
small
vessel
stents
was
established
by
measuring
in-stent
late
loss
by
QCA
at
9-months
(primary
endpoint)
as
well
as
TLF
at
12
months
as
compared
to
a
performance
goal
(secondary
endpoint,
based
on
TAXUS
Express
Stent
results
from
the
TAXUS
IV
and
TAXUS
V
trials).
The
study
success
criteria
are
met
when
mean
in-stent
late
loss
at
9
months
post-index
procedure
for
the
ION
Stent group
is
shown
to be
superior
to
that
of
the
historical
control
and
the
12-month
TLF
rate
for
the
ION
Stent
group
is
shown
to
be
less
than
the
performance
goal.
The
analyses
showed
that
the
ION
small
vessel
stents
were
superior
(p-value
<0.0001)
to
both
the
in-stent
late
loss
historical
control
at
9
months
and
TLF
rate
at
12
months.
C.
Overall
Conclusions
The
clinical
testing
conducted
demonstrated
that
the
IONTM
Paclitaxel-Eluting
Coronary
Stent
Systems
(Monorail
and
Over-The-Wire)
provides
a
reasonable
assurance
of
safety
and
effectiveness
when
used
as
indicated
in
accordance
with
the
instructions
for use.
XII.
CDRH
DECISION
CDRH
issued
an
approval
order
on
April
22, 2011.
The final
conditions
of
approval
cited
in
the
approval
order
are
described
below.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
54

1.
BSC
should
collect
and
report
to
the
Agency
on
an
annual
basis,
clinical
outcomes
through
5
years
post-procedure
on
at
least
80%
of
patients
enrolled
(excluding
those
discontinued
due
to
death)
in
the
PERSEUS
Workhorse
and
PERSEUS
Small
Vessel
studies.
When
appropriate
or
as
requested
by
FDA,
BSC
should
submit
PMA
supplements
requesting
approval
to
update
your
Instructions
for
Use
(IFU)
to
include
these
data.
2.
BSC
should
collect
clinical
data
on
the
implantation
of
the
PMA-approved,
commercially
distributed
ION
product
in
the
U.S.
The
trial
should
be
statistically
powered
to
evaluate
the
annual
rates
of
stent
thrombosis,
and
the
rate
of
cardiac
death
plus
myocardial infarction
(MI)
through
five
years
in
patients treated
with
the
ION
product
according
to
its
labeled
indications.
These data
are
needed
to
evaluate
whether
the
rate
of
stent
thrombosis
plateaus
or
increases
over
time,
and
to
evaluate
the
impact
of
stent
thrombosis
on
rates
of
cardiac death
and
MI.
These
data
are
also
needed
to
evaluate
the
potential
for
rare
adverse events related
to
the
drug substance
and/or
drug
carrier
that could
not
be
detected
in
your
initial
clinical
trials.
BSC
has
proposed
collecting
these data
from
at
least
2,500
patients
at
up
to
50
U.S.
sites
in
the
ION
Postmarket
Registry.
FDA
agrees
that
the
registry
protocol
submitted
in
the
original
PMA
is
acceptable.
Please
provide
progress
reports
at
6,
12,
18,
and
24
months
and
annually
thereafter
through
5
years
with
data
from
your
U.S.
registry.
When
appropriate
or
as
requested
by
FDA,
BSC
should
submit
PMA
supplements
requesting
approval
to
update your
IFU
to
include these
data.
Please
note
that
if
subsequent
data analyses identify
areas
of
significant
off-label
use,
the
firm
should
submit
an
IDE
to
conduct
an
appropriate
study
to
evaluate
the
off-label
use.
3.
The
issue
of
the
optimal
duration
of
dual
antiplatelet
therapy following
PCI
with
drug-eluting
stents
(DES)
remains
a
critical
question
that
is
currently
being studied
in
the
DAPT
trial.
FDA
acknowledges
that
BSC
is
participating
in
this
trial
to
address
a
condition
of
approval
for
the
Taxus
Liberte
DES
(P060008).
As
the
duration
of
dual
antiplatelet
therapy
is
also
relevant
for
the
ION
Paclitaxel-Eluting
Coronary
Stent, the
sponsor
should
fulfill
their commitment
to
the
condition
of
PMA
approval
for
P060008.
When
appropriate
or
as
requested
by
FDA,
BSC
should
submit
PMA
supplements
to
the
ION
PMA
(P100023)
requesting
approval
to
update
their
IFU
to
include
the
data
collected
in
the
DAPT
trial.
If
BSC
does not
fulfill
the
condition
of
approval
for
P060008,
they
should
conduct
or
participate
in
a
separate clinical
trial
that
will
develop
data
to
study
the
duration
of
dual
antiplatelet therapy
following
implantation
of
the
ION
DES
and
subsequently
submit PMA
supplements
to
this
PMA requesting
approval
to
include
these data
in
an
IFU
update.
4.
BSC
has agreed
to
provide
annual
stability
data
for
their
Maple
Grove
manufacturing
site
as
part
of
their
annual
reports.
The
applicant's
manufacturing
facilities
were
inspected
and
found
to be
in
compliance
with
the
device
Quality System
(QS)
regulation
(21
CFR
820).
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
55

XIII. APPROVAL
SPECIFICATIONS
Directions
for
use:
See
device
labeling.
Hazards
to
Health
from
Use
of
the
Device:
See
Indications,
Contraindications,
Warnings,
Precautions,
and
Adverse Events
in
the
device
labeling.
Post-approval
Requirements
and
Restrictions:
See
approval
order.
XIV.
REFERENCES
1.
Allocco
DJ,
Cannon
LA,
Britt
A,
Heil
JE,
Nersesov
A,
Wehrenberg
S,
Dawkins
KD,
Kereiakes
DJ.
A
prospective
evaluation
of
the
safety
and
efficacy
of
the
TAXUS
Element paclitaxel-eluting
coronary
stent system
for the
treatment
of
de
novo
coronary
artery
lesions:
Design
and
statistical
methods
of
the
PERSEUS
clinical
program.
Trials.
2010;11(1):1
2.
King
SB,
3rd,
Smith
SC, Jr.,
Hirshfeld
JW, Jr.,
Jacobs
AK,
Morrison
DA,
Williams
DO,
Feldman
TE,
Kern
MJ,
O'Neill
WW,
Schaff
HV,
Whitlow
PL,
Adams
CD,
Anderson
JL,
Buller
CE,
Creager
MA,
Ettinger
SM,
Halperin
JL,
Hunt
SA,
Krumholz
HM,
Kushner
FG,
Lytle
BW,
Nishimura
R,
Page
RL,
Riegel
B,
Tarkington
LG,
Yahcy
CW.
2007
Focused
Update
of
the
ACC/AHA/SCAI
2005
Guideline Update
for
Percutaneous
Coronary
Intervention:
a
report
of
the
American College
of
Cardiology/American
Heart
Association
Task
Force on
Practice
Guidelines:
2007
Writing
Group
to
Review New
Evidence
and
Update
the
ACC/AHA/SCAI
2005
Guideline Update for
Percutaneous
Coronary Intervention, Writing
on
Behalf
of
the
2005
Writing
Committee. Circulation.
2008;
117(2):261-95.
3.
From
Table
1
in
Popma,
JJ,
Tiroch,
K,
Almonacid,
A,
Cohen,
SA,
Kandzari,
DE,
and
Leon,
MB.
A
Qualitative
and
Quantitative
Angiographic
Analysis
of
Stent
Fracture
Late
Following
Sirolimus-Eluting
Stent
Implantation.
Am
J
Cardiol
103:
923-929,
2009.
4.
Cutlip
DE,
Windecker
S,
Mehran
R,
et
al.
Clinical
End
Points
in
Coronary
Stent
Trials:
A
Case for
Standardized Definitions. Circulation.
2007;115(17):2344-2351.
5.
Shaw
LJ,
Bairey
Merz
CN,
Pepine
CJ,
et
al.
Insights
From
the
NHLBI-Sponsored
Women's
Ischemia
Syndrome
Evaluation
(WISE)
Study:
Part
1:
Gender Differences
in
Traditional
and
Novel
Risk Factors,
Symptom
Evaluation,
and
Gender-Optimized
Diagnostic
Strategies.
J
Am
Coll
Cardiol
2006
47:
S4-20.
6.
Lansky
AJ,
Costa
RA,
Mooney
M,
et
al.
Gender-Based
Outcomes After
Paclitaxel-
Eluting Stent
Implantation
in
Subjects With Coronary
Artery
Disease.
J
Am
Coll
Cardiol
2005
45:
1180-5.
PMA
P100023:
FDA
Summary
of
Safety
and
Effectiveness
Data
Page
56
